Dispersion polymerization induced self-assembly (pisa) techniques for the fabrication of polymeric nanoparticles for biomedical applications
Received: 16-Dec-2022, Manuscript No. PULNL-22-5916; Editor assigned: 19-Dec-2022, Pre QC No. PULNL-22-5916 (PQ); Accepted Date: Dec 28, 2022; Reviewed: 21-Dec-2022 QC No. PULNL-22-5916 (Q); Revised: 26-Dec-2022, Manuscript No. PULNL-22-5916 (R); Published: 28-Jan-2023, DOI: 10.37532. pulnl.23.8 (1) 01-015
Citation: Akala E.O. Dispersion polymerization induced self-assembly (pisa) techniques for the fabrication of polymeric nanoparticles for biomedical applications. Nanotechnol. Lett. 2022;8(1):01-15
This open-access article is distributed under the terms of the Creative Commons Attribution Non-Commercial License (CC BY-NC) (http://creativecommons.org/licenses/by-nc/4.0/), which permits reuse, distribution and reproduction of the article, provided that the original work is properly cited and the reuse is restricted to noncommercial purposes. For commercial reuse, contact reprints@pulsus.com
Abstract
Nanoparticles offer several advantages in drug delivery. The progress in the development of nanoparticles for biomedical applications has moved from the first generation nanoparticles to the fifth generation nanoparticles and the transitions reflect their increasing versatility in biomedical applications. Polymeric nanoparticles are prepared mainly by two methods: dispersion of preformed polymers and in situ polymerization of monomers and macromonomers. Polymerization induced self-assembly (PISA) for the fabrication of nanoparticles is believed to be a better strategy than nanoparticle fabrication from preformed polymers (ease of tethering targeting ligands to the corona of the nanoparticles and unlike PISA, creation of nanostructures via self-assembly of block copolymers is performed in low concentrations. Dispersion polymerization involves one-pot synthesis of nanoparticles. RDRP processes such as atom transfer radical polymerization, reversible addition-fragmentation chain transfer polymerization and nitroxide mediated polymerization have revolutionized polymer synthesis by providing polymer chemists with powerful tools that enable control over architecture, composition and chain length distributions. The technique for the fabrication of nanoparticles by dispersion polymerization (PISA) at ambient temperature was described with examples from our laboratory involving organic redox initiated polymerization using the FDA approved biodegradable polymers. Computer optimization is useful in understanding the factors that ensure optimized properties of drug-loaded nanoparticles
Keywords
Nanotechnology; Nanoparticles; Dispersion polymerization, Computer optimization; Polymerization induced self-assembly; Biodegradable polymer; cancer chemotherapy; Polymeric drug; Delivery system; Quality by design
Introduction
Nanoparticles are submicron (< 1 μm) colloidal systems which can be fabricated from varied and diverse materials in a variety of compositions, including quantum dots (QDs), polymers, gold, paramagnetic iron, etc [1]. Recent advances in the field of nanotechnology have made nanoparticles very promising in the delivery and targeting of bioactive agents, drug discovery and diagnostics.
In fact, nanotechnology has been described as one of the key technologies of the 21st century [2]. The progress in the development of nanoparticles for biomedical applications has moved from the first generation nanoparticles— mainly suitable for liver targeting, as they are captured in the liver by the reticuloendothelial system (RES), also known as mononuclear phagocyte system (MPS); into the second generation, stealth nanoparticles: the nanoparticle surface is decorated or tagged with water soluble polymers, such as polyethylene glycol (PEG) or N-(2-Hydroxypropyl)methacrylamide (HPMA) for long systemic circulation and passive targeting (sequestration of the nanoparticles into the leaky vasculature of the tumor blood vessel, followed by their retention—enhanced permeability and retention (EPR) effect); to the third generation nanoparticles, with targeting moiety. The nanoparticle surface is decorated with a ligand specific for the antigen or receptor expressed on the surface of the tumor/pathological cells with a view to targeting the biophase (site of action), thereby achieving target specific delivery, reducing or eliminating off-target toxicity, and reducing the therapeutic dose[3-5]. The fourth generation nanoparticles have been dubbed theranostic: multifunctional nanoparticles which allow for a combination of diagnostic agent with a therapeutic agent and a reporter of therapeutic efficacy in the same nanodevice package [6].
Advantages Of Nanoparticles
Nanoparticles offer several advantages in drug delivery due to their unique characteristics. Some of these characteristics and advantages include the following: potential for functionalization for enhanced drug-carrying capacity [7], tissue or organ specific transport and delivery [7,8] reduction in administered dose and toxicity [9], the ability to carry and deliver multiple classes of diagnostics and therapeutic agents loaded within a nanoparticle, which would then exert their various effects in a controlled manner [7,10] and reduction in the frequency of administration [9].
The capability of nanoparticles to bear multiple therapeutic agents would make it easier to administer drugs in combination without having to increase the frequency of administration. Thus, therapeutic agents belonging to different classes with different physicochemical properties can be combined within the same nanoparticle system to achieve the desired therapeutic goal. Combination therapy using nanoparticle formulations provides certain advantages over combining the free drugs for therapy. The controlled release feature offered by nanoparticle systems can normalize the pharmacokinetics, biodistribution, and stability of drugs that possess very different chemical properties that would independently have produced contrasting pharmacological behaviors. These long-circulating formulations are capable of continuous release of drugs at controlled ratios or permit independent modification of release rates of each drug in ways that would not be achievable with conventional formulations of free drug which are rapidly cleared from the system [11].
Varying biodistribution/pharmacokinetics of combination drugs through cocktail administration has been attributed to their ineffectiveness in the clinic [12]. The problem is being solved by nanotechnology platform for drug delivery. The unique ability of multifunctional therapeutic nanoparticles to provide site-specific. Tumor targeting, improve the solubility of anticancer drugs, synchronize the disposition (pharmacokinetics) of encapsulated drugs (drug combination), overcome drug resistance and enhance anticancer activity of therapeutic drugs (concurrent chemotherapy with trastuzumab or pertuzumab is more effective than sequential use of these agents) represents an important innovation in drug delivery system [13-22]. Nanoparticles are also important tools for imaging and diagnosis, aside from therapy or treatment [1] . The most commonly used imaging techniques are positron emission tomography (PET), single photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and various optical imaging techniques (bioluminescence and fluorescence) that have high sensitivity. Among these techniques, MRI is the most commonly studied technique and a considerable amount of research has been devoted to the use of magnetic particles as contrast agents. Particles of gadolinium, iron oxides, gold, silver and other metals are currently being investigated [23,24].
Though MRI is a noninvasive technique routinely used clinically for diagnostic imaging, it is believed that magnetic resonance sensitivity is significantly low in comparison with optical and nuclear imaging [25]. To improve MRI to the level where detection of molecular markers becomes possible, special contrast agents which significantly amplify the MR signals are often used. Advances in the application of MR methods for breast cancer research have become possible from the development of contrast agents that generate receptor-target or molecular-target contrast. The noninvasive MRI has a big role in functional imaging: cancer diagnosis and staging, in which the contrast agents in nanoparticle core have found applications in defining tumor margins, characterizing tumor perfusion and identifying tumor-bearing lymph nodes [26,27].
Fabrication Of Polymeric Nanoparticles
Polymeric nanoparticles can be prepared mainly by two methods:
(i) dispersion of preformed polymers and
(ii) in situ polymerization of monomers, crosslinking agents and macromonomers.
Fabrication of polymeric nanoparticles from dispersion of preformed polymers
Self-assembly of block copolymers in a selective solvent (traditional post polymerization solvent switch approach) is an important strategy for preparation of polymer materials in nanoscale, and a broad range of intricate, biomimetic nanomaterials, including spherical micelles, nanorods, vesicles, tubes and donuts, etc. have been created. It utilizes block copolymers consisting of a soluble block and an insoluble block. The method has been used successfully in the fabrication of polymeric nanoparticles. These nanostructural materials show potential applications in varied and diverse fields, such as in catalysis, biomedical, food and cosmetic industries [28]. Examples of the methods used are as follows: emulsification solvent evaporation, emulsification solvent double emulsion method, nanoprecipitation, microphase-inversion, salting out, dialysis, and supercritical fluid technology
Disadvantages of the method include:
• It is very difficult to tether targeting ligands, like mAbs, to the corona/ surface of the nanoparticles for biorecognition events. Any attempt to modify the surface of nanoparticles fabricated by dispersion of preformed polymers often results in a substantial loss ofencapsulated bioactive agents, contrast agents for imaging or other materials.
• Generally, creation of nanostructures via self-assembly of block copolymers is performed in low concentrations and involves multiple steps which prevents its commercialization and further applications. The preparation of multimorphologies via polymerization-induced selfassembly (to be discussed below) and morphology transition can be conducted with the monomer concentration as high as 500 mg mL-1 which is not possible with selfassembly of block copolymers [28] in a selective solvent, in which 1 mg mL-1 of copolymer is used generally.
Fabrication of polymeric nanoparticles by in situ polymerization
In situ co-polymerization of monomers/macromonomers, including crosslinkers, is another method for the fabrication of polymeric nanoparticles; it is also called polymerization-induced self-assembly (PISA). It is a chemical reaction that drives a physical polymer self-assembly process (polymerizationinduced self-assembly (PISA) combines block copolymer synthesis and nanoparticle formation efficiently at high polymer concentrations). Various nanoparticle morphologies such as spheres, worms, and vesicles can be prepared readily in polar and nonpolar media. The method allows one-pot synthesis of nanoparticles. It offers many advantages such as easy functionalization of the polymeric nanoparticles’ surface (needed to modify the biodistribution of the nanoparticles for long blood circulation by avoiding capture by the reticuloendothelial system (passive targeting), site specific uptake in cells (active targeting) by tethering a ligand to nanoparticle surface that can achieve biorecognition by virtue of the receptors expressed on the surface of cells (e.g., cancer cells), incorporation of pH-sensitive monomers and crosslinking agents for controlled/sustained drug release [19-23,29]. Theranostics nanoparticles (multifunctional nanoscale devices which allow for a combination of diagnostic agent with a therapeutic agent and even a reporter of therapeutic efficacy in the same nanodevice package) can be easily made by in situ polymerization method [30]. All these advantages of in situ polymerization derive from the possibility of simultaneous encapsulation of relevant hydrophobic/ hydrophilic drugs, contrast agent, nucleic acids, fluorochromes and, by copolymerization, adding surface functionalities in one batch process, without further modifications. In nanoparticle fabrication by dispersion of preformed polymers in selective solvents, any attempt to modify the surface of nanoparticles often results in a substantial loss of encapsulated drugs or other materials. Among the techniques available for in situ polymerization for the fabrication of nanoparticles are emulsion polymerization, microemulsion polymerization, miniemulsion polymerization, dispersion polymerization, and suspension polymerization[18-22,29,31,32].
Dispersion Polymerization Techniques For The Fabrication of Polymeric Nanoparticles For Biomedical Applications
Dispersion polymerization occurs in the presence of a suitable polymeric stabilizer soluble in the reaction medium, which is adsorbed on the surface of particles where it lowers the surface free energy and functions as a steric stabilizer [18-22,33]. The starting reaction mixture is a clear, singlephase solution (a homogeneous solution of monomers, initiator, drug and stabilizer) with particles forming by precipitation of growing polymer chains in the presence of a suitable steric stabilizer. Consequently, the solvent medium becomes a dispersion medium [34,35]. Dispersion polymerization has many advantages over other methods of polymeric particle rapid reaction rate by simple free-radical reactionpreparation. These advantages include:
i. Rapid reaction rate by simple free-radical reaction
ii. Elimination of toxic organic solvents and non-use of surfactants which makes it especially applicable to biomedical applications.
iii. Production of spherical monodisperse particles in a single step (simultaneous encapsulation of bioactive agents during polymerization) and achievement of nanoparticle preparation of multi-morphologies via polymerization-induced self-assembly; morphology transition can be conducted with the monomer concentration as high as 500 mg mL-1
iv. It can be carried out at room temperature by using appropriate initiators
v. Single homogenous phase at the start of the polymerization reaction when compared to the multiple phases present in an emulsion polymerization process
vi. It is possible to add surface functionalities in one-batch process (one-pot synthesis) without further modifications compared to nanoparticle fabrication by dispersion of preformed polymers [18,22,28,33]
Dispersion polymerization in the fabrication of stealth nanoparticles is noteworthy. It has been reported that a few physical protocols have been adopted to coat nanoparticle with PEG but these procedures entail the risk of polymer desorption in the blood with consequent loss of the beneficial contribution of the polymer. In order to overcome this problem, covalent PEG conjugation protocols have been developed for biodegradable nanoparticles with PEG covalently bound to the surface and have been produced using PEG derivatives of poly(lactic acid), poly(lactic acid-coglycolic acid) or poly(alkylcyanoacrylates). The nanoparticles are often prepared by dispersion polymerization in various types of media. These procedures allow the PEG orientation toward the water phase, while the biodegradable hydrophobic polymer fraction is physically entangled in the inner nanoparticle matrix [36].
• Reversible-deactivation radical polymerization (RDRP)
RDRP processes (also known as living or controlled radical polymerization), such as atom transfer radical polymerization (ATRP), reversible additionfragmentation chain transfer (RAFT) polymerization and nitroxide mediated polymerization (NMP) have revolutionized polymer synthesis by providing polymer chemists with powerful tools that enable control over architecture, composition and chain length distributions. A living radical polymerization (LRP) is a free radical polymerization that aims at displaying living character, (i.e., does not terminate or transfer and is able to continue polymerization once the initial feed is exhausted by addition of more monomer). Termination reactions are inherent to a radical process, and modern LRP techniques seek to minimize such reactions, therefore providing control over the molecular weight and the molecular weight distribution of a polymeric material. In addition, the LRP techniques allow compatibility with a wide range of monomers, tolerance of many functionalities, and facile reaction conditions. The control of molecular weight and molecular weight distribution has enabled access to complex architectures and site-specific functionality that were previously impossible to achieve via traditional free radical polymerizations [37].
The user-friendly nature of these procedures have allowed RDRPderived polymers to be used in the construction of advanced materials with unique and enhanced properties. RDRP has been widely explored for the systematic design and synthesis of biomaterials largely due to the enhanced control over polymer structure and mild reaction conditions. In particular, polymers synthesized through RDRP have been applied in three main areas to provide biomaterials with specific and enhanced properties, namely, i) the conjugation of synthetic polymers to biomacromolecules such as peptides, proteins and siRNA, ii) the development of functional polymeric nanoparticles for the transport of therapeutic and imaging agents and iii) the development of bioactive polymers that can trigger biological responses. These three areas all take advantage of RDRP to precisely control the polymer architectures and molecular weights [38,39]. RDRP has been successfully employed to prepare a broad range of amphiphilic copolymers with various functionality that undergo self-assembly to form supramolecular structures with morphologies ranging from spheres to vesicles and higher ordered structures. These polymeric nanoparticles can provide a vehicle for the delivery of therapeutic agents and can be decorated with targeting moieties[40,41] .
Reversible-deactivation radical polymerization (RDRP) has been successfully implemented in dispersed media, and one of the most significant achievements is the development of synthetic routes allowing the production of block copolymer nano-objects. This approach, known as polymerization-induced selfassembly (PISA), takes advantage of the chainend reactivity of solvophilic macromolecules obtained by RDRP for the polymerization of a second monomer in a suitable solvent. The growth of the second block, insoluble in the polymerization medium, leads to the formation of block copolymers that self-assemble into nanoparticles[42]. PISA can be performed in dispersion polymerization conditions in which the monomers are soluble. Under optimized conditions, PISA can directly produce the same self-assembled morphologies (spheres, rods, fibers, vesicles) previously obtained by the solvent-displacement method using preformed block copolymers, but at much higher solids contents (up to 40– 50%) and with significantly less experimental effort. The great majority of the PISA systems reported so far rely on dispersion polymerization, in which particle morphology is generally more easily tuned [42]. Other RDRP techniques have also successfully been implemented in the past, such as atom transfer radical polymerization (ATRP), nitroxide-mediated polymerization (NMP), organotellurium mediated radical polymerization (TERP), organometallic mediated radical polymerization (OMRP), and iodine transfer polymerization (ITP). These techniques, however, present various drawbacks compared to RAFT polymerization, and remain much less exploited in the field of PISA. Very recently, it was shown that PISA is not only feasible with RDRP, but that the principles also hold for other polymerization mechanisms, in particular ring-opening metathesis polymerization (ROMP) [42] .
• Reversible addition-fragmentation chain transfer (RAFT) method.
RAFT enables control over polymerization of most monomers available to free radical polymerization. Furthermore, RAFT also offers some benefits when considering monomers that are challenging to polymerize by conventional free radical polymerization. A key requirement in RAFT, which is very different from other RDRP systems, is the use of a radical initiator[43]. The RAFT process is a simple modification of a conventional free radical polymerization process by substituting a traditional chain transfer agent with a RAFT agent. Block copolymers of the type AB are one of the key products achievable via RAFT, and they are produced by sequential addition of a monomer B to a macro-RAFT agent produced by the polymerization of monomer A, mediated by the RAFT agent [43]. RAFT has been described as the most well-established PISA method. So far, it is the most versatile (in terms of monomer and solvent compatibility) and reliable polymerization technique for PISA [44] . PISA is based on the chain extension of an initial soluble precursor block, which acts as a steric stabilizer, with a second insoluble polymer block that forms the nanoparticle core in situ. This polymerization reaction triggers diblock copolymer self-assembly once a sufficiently high degree of polymerization (DP) of the coreforming block is attained. PISA is efficient because polymer synthesis and assembly occur simultaneously. Furthermore, PISA can be performed at a range of final polymer concentrations (5–50% w/w) [45]. RAFT allows nanoparticle cross-linking and can be performed in polar solvents, such as water; various organic solvents and alcohols; and in nonpolar solvents, such as n-alkanes and mineral oil. RAFT-PISA can also be performed in ionic liquids and supercritical CO2. A range of morphologies can be obtained when using RAFT dispersion polymerization. An example of an aqueous RAFT dispersion polymerization is the poly(glycerol monomethacrylate)-blockpoly(2-hydroxypropyl methacrylate) (PGMA-b-PHPMA), where PGMA forms the soluble stabilizer block, and PHPMA forms the insoluble nanoparticle core [46-48]. Nonpolar RAFT-PISA formulations prepared by RAFT exist. A good example of such formulations is the poly(lauryl methacrylate)-block-poly(benzyl methacrylate) (PLMA-PBMA) composition in various n-alkanes where the PLMA blocks act as an oil-soluble stabilizer and the PBMA blocks form the insoluble nanoparticle core [49] Polar and nonpolar RAFT-PISA formulations have similar characteristics in terms of in situ morphological evolution. Generally, this morphological evolution proceeds from dissolved polymer chains to spheres, to worms, to vesicles[50]. It is known that RAFT-PISA has allowed the preparation of nanoparticles that are suitable in various medical applications, including long-term stem cell storage and drug delivery [51,52] .
Some disadvantages have been highlighted for RAFT. RAFT-PISA is a formidable technique for the in situ preparation of dispersed nanoparticles in various media. However, this technique has its limitations. An aspect that can be considered a disadvantage of PISA formulations is that they generally require thermal initiators (reaction temperatures are ∼ 70–90°C), though light-controlled radical polymerization reactions at ambient temperatures have been reported [53]. Judiciously chosen photoinitiators allow RAFT polymerization reactions in the presence of light; while the absence of light pauses the polymerization reaction. Such initiator systems have been investigated for RAFT-PISA [53-57]. Nanoparticles obtained from RAFT-PISA contain sulfur-containing polymer end groups that are located within the nanoparticle cores. These are potentially harmful and cause intrinsically colored dispersions. Furthermore, these polymer end groups give an undesired odor to the polymers. Fortunately, convenient techniques have been developed to degrade the polymer chain ends from nanoparticles prepared via RAFT-PISA in polar and nonpolar media. However, these end groups’ stability is desired during RAFT-PISA to maintain control over the polymerization reaction. For this reason, aqueous RAFT-PISA is generally performed under acidic or neutral conditions: RAFT end groups are susceptible to hydrolysis above pH 7 [38]. These limitations can be avoided when using polymerization techniques that avoid sulfurcontaining end groups, as discussed in the rest of this review
• Atom transfer radical polymerization (ATRP)
A controlled living radical polymerization methodthat has competed with RAFT polymerization over the years is ATRP. There are several reasons why ATRP is less suitable for PISA than RAFT. For example, ATRP utilizes a copper catalyst, which forms an undesired potential toxic impurity in the polymer product. For this reason, nanoparticles obtained from ATRP-PISA are less suitable for biomedical/pharmaceutical applications. Copper removal is possible; for example, silica column chromatography could be used after nanoparticle cross-linking. Another recent study described a method that utilizes a “Cu scavenger”, followed by filtration, on a polymer solution. Such procedures are demanding and would require cross-linked nanoparticles. There are other disadvantages of ATRP-PISA which arise from using a metal catalyst. For example, ATRP metal complexes are often vulnerable to oxidation. Fortunately, different ATRP methods have been developed to improve upon this limitation and to allow this polymerization reaction with low copper concentrations [44]. A recent example of ATRP-PISA was reported by Matyjaszewski and co-workers [58]. ATRP-PISA was fabricated with a reduced copper concentration. This was achieved by employing the ICAR-ATRP method. A polyoligo(ethylene glycol) methyl ether methacrylate] (POEOMA) macroinitiator was chain extended with PBMA in ethanol at room temperature and at 65°C. The effects of catalyst concentration, radical initiators, target PBMA DP, solids content, and temperature were investigated for this dispersion polymerization. High monomer conversions and relatively narrow MWDs were obtained. Only spheres are formed at low polymer concentrations. Spheres with diameters of ∼300 nm and worms were observed at higher final polymer concentrations. In contrast to the samples obtained at 65°C, the worms obtained at room temperature had short fractal-type connected-bead morphology, which suggests that these elongated structures are formed by a sphere–sphere fusion process[58]. Another recent ATRP-PISA contribution was reported by Zetterlund and co-workers [59]. This formulation comprised chain extension of poly(dimethylsiloxane) and BMA in supercritical CO2. TEM images of the poly(dimethylsiloxane)- block-poly(benzylmethacrylate) (PDMA-b- PBMA) nanoparticles suggested the presence of spheres, worms, and possibly, vesicles.
• Nitroxide-mediated polymerization (NMP): NMP, RAFT and ATRP are being used in the synthesis of well-defined homo-, gradient, diblock, triblock, and star polymers and other architectures, including microgels and polymer brushes. New materials that have the potential of revolutionizing a large part of the polymer industry are beginning to appear [60]. NMP proceeds via reversible homolytic dissociation of terminal alkoxyamine groups (reversible thermal homolysis of alkoxyamines into alkyl and nitroxyl radicals). Investigations have led to the development of variants of NMP which have been used for the investigation of materials prepared using PISA techniques [61]. Macroalkoxyamine has been used to play the role of both control agent and stabilizer during the polymerization. This approach has been used with success for various systems in dispersed media leading to the formation of complex morphologies [62]. NMP has shown promising results in terms of PISA. Charleux and co-workers reported spherical nanoparticles formation from the chain extension of poly(sodium acrylate) with styrene and n-butyl acrylate (BA) in water at 20% w/w. These dispersion polymerizations yielded spherical nanoparticles [63,64]. Later, Charleux and co-workers showed that NMP-PISA could be used to prepare crosslinked and uncross-linked poly(sodium acrylate)- block-poly(N,Ndiethylacrylamide) spheres [65.] Furthermore, Charleux and co-workers reported in 2009 that the poly(sodium acrylate)-block-poly(4-vinylpyridine) composition allows self-assembly into well defined spheres, worms, and vesicles [66].
Organic Redox Initiated Dispersion Polymerization Technique for the Fabrication of Nanoparticles by PISA at Ambient Temperature
• Organic redox initiator system for the fabrication of nanoparticles (PISA of nano-objects)
Our laboratory has been involved in the application of in-situ dispersion polymerization technique at ambient temperature involving redox initiator system for the fabrication of core-shell nanoparticles (PISA of nanoobjects) [14-19,21,22]. The uniqueness of in-situ dispersion polymerization at ambient temperature involving redox initiator system for the fabrication of core-shell nanoparticles are as follows: Nanoparticle fabrication at ambient temperature is suitable for thermolabile bioactive agents like peptide and proteins (especially monoclonal antibodies); it is surfactant free thereby obviating the problems associated with the use of surfactants in injectable liquid preparations; it is a one-pot synthesis.
Homolytic breakage of covalent bonds in initiator molecules by absorption of energy and the transfer of electrons from atoms with unpaired electrons followed by bond dissociation in the acceptor molecule have been reported as primary radical generation mechanisms in free radical initiation reactions. The most effective of the electron transfer reactions are redox reactions capable of generating free radicals and initiating polymerization reactions under mild conditions [67]. Redox reactions have notable advantages over thermal homolytic degradation of the covalent bond of initiator molecules in PISA (nanoparticle synthesis): (a) a higher decomposition rate constant of the initiator molecule leading to a short induction period [67,68]. (b) a lower activation energy that promotes milder polymerization conditions which is important in the encapsulation of thermolabile products like peptides and proteins [14] (c) reduction in side chain reactions leading to high molecular weight polymers with improved yield [67] .The most commonly used redox initiator system is the organic peroxide/amine system and the peroxide most commonly used is benzoyl peroxide (BPO). The free radical is generated by the decomposition of BPO activated by tertiary amines [68,69]. A number of tertiary amines have been used as activators including N-phenyldiethanolamine [14]. Vazquez et al [70], in their review, made a list of tertiary amines used as activators of BPO and also gave a good review of the toxicity of commonly used tertiary amines. The dispersion polymerization reactions for the fabrication of PISA (nanoparticles) in our laboratory have been based on redox initiator system comprising BPO and N-phenyldiethanolamine (NPDEA). The radical production is as shown in (Scheme 1) below [1,68] .

Scheme 1: Mechanism of generation of free radical at ambient temperature in the BPO/NPDEA redox initiator system
• Polylactide and poly-ɛ-caprolactone biodegradable block materials for core-shell nanoparticles (PISA of nano-objects) in our laboratory
Polymers that have been investigated for the fabrication of polymeric nanoparticles include natural macromolecules (biopolymers) and synthetic polymers [5]. The major technique in the design of biomedical polymeric nano-devices is based on physical or chemical combination of drug(s) with polymers. Given the complexities of natural polymers, research efforts are shifting towards synthetic polymers, because they can be synthesized reproducibly and predictably, thereby permitting the selection of materials of formulation with uniform and controlled composition. Certain requirements are expected of polymers for the fabrication of drug delivery devices in general, including polymeric core-shell nanostructures [71,72].
a) The drug should show good diffusion and solubility characteristics in the polymer to provide the desired release control.
b) The polymer must be compatible with the host environment (e.g., not toxic or antagonistic in medical applications).
c) The polymer must be stable (should not degrade or change undesirably).
d) The polymer must be compatible with the bioactive agent (no undesirable reactions or physical interactions).
e) The polymer must exhibit optimum mechanical properties. The polymer must be easily manufactured, fabricated into desired shape, easily sterilized and inexpensive.
f) The polymeric material biocompatibility has to be defined only in the precise context of material use: a polymer may be biocompatible in one application but not biocompatible in another (should be compatible with blood if contact with blood is desirable; should be compatible with the tissue in question if not blood).
g) Polymeric nanoparticles are of the same size as biological entities; consequently, they can readily interact with biomolecules on both the cell surface and within the cell.
Polymer molecular weight distribution, charge, hydrophobicity, etc., have a profound effect on the polymer biocompatibility. The molecular weight of non-biodegradable polymers should be about 40 kDa to ensure renal elimination. (j). It is believed that polycations are significantly more toxic than water soluble natural polymers and polyanions; however, a few polycation-based systems (chitosan for example) have been developed and tested in clinical applications
The most commonly used synthetic polymers for the fabrication of nanoparticles are PLA, PLGA, poly(glycolic acid) (PGA), poly(ε-caprolactone) (PCL) and poly (β-hydroxybutyrate) (PHB). Belonging to the family of polyesters, these polymers are known to exhibit adequate biodegradability and biocompatibilty. Under physiological conditions, polyesters are generally degraded by hydrolysis into products which are well tolerated by various tissues. For example, the degradation products from PLA, PGA, and PLGA, namely glycolic acid and lactic acid, are physiological substances easily eliminated through the Krebs cycle [3]. The successful use of polymers of lactic acid and glycolic acid (PLA, PGA, and PLGA) as biodegradable drug delivery systems and as biodegradable sutures led naturally to an evaluation of other aliphatic polyesters, and to the discovery of the degradability of PCL in vivo [17,73]. Several studies using copolymers of poly-caprolactone have shown that it is biocompatible. Poly-caprolactone undergoes hydrolytic degradation to give an intermediate which produces 6-hydroxycaproic acid. 6-Hydroxycaproic acid is broken down to acetyl-CoA units via β-oxidation (fatty acid metabolism) for further degradation via the Krebs cycle. Thus PCL is degradable to products that are physiologically metabolized by the body. Among the United States Food and Drug Administration approved polyesters such as PLA, PGA and PLGA, PCL possesses unique properties, such as higher hydrophobicity and neutral biodegradation end products, which do not disturb the pH balance of the degradation medium [74,75]. Over the years, many types of drug delivery systems, including nanoparticles, have been developed using PCL as polymeric material [5]. Aside from poly (esters), other classes of biodegradable synthetic polymers that have been used in the fabrication of nanoparticles are as follows: polyorthoesters, po lyanhydrides,polycarbonates,polyphosphazenes , polyphosphoesters, and polyamides. Some of the polymers/copolymers that have been used in the fabrication of nanoparticles are shown in the literature[1]
i. Poly-ɛ-caprolactone macromonomer
Polyester macromonomers are linear macromonomers carrying polymerizable functional groups at their chain ends. The end-capping of poly-ɛ-caprolactone macromonomers can be achieved by polymerizing ɛcaprolactone using an initiator (e.g. aluminium isopropoxide) followed by esterification of hydroxyl end group by a suitable compound (e.g. methacrylic acid), which involves a two-step process. Alternatively, ring opening polymerization of ɛ-caprolactone can be done in the presence of an initiator carrying the required functional group. Consequently, following polymerization, one chain end bears an hydroxyl group while the other is capped with the functional group associated with the initiator (one-step process). In our work, methacrylate end functionalized poly-ɛcaprolactone (P(CLHEMA)) was synthesized by a modified published method using hydroxylethylmethacrylate as the initiator (one-step process as shown in Scheme 2) [5,17,21,22,76,77]

Scheme 2: Ring-opening polymerization scheme for the synthesis of poly(-ɛ-caprolactone) macromonomer [5]
TABLE 1. Properties of four different batches of poly-e- caprolactone-HEMA macromonomer
| Molar ratio Caprolactone /HEMA (Feed Composition) | Mn (1H NMR) | Mw (GPC) | Polydispersity Index (PDIpolymer) | HEMA (Mole in Feed Composition | Mol% HEMA ( 1H NMR) in Macromonomer |
|---|---|---|---|---|---|
| 14.96 | 1916 | 6445 | 2.13 | 0.0077 | 5.5 |
| 7.53 | 1455 | 3735 | 1.64 | 0.0153 | 7.7 |
| 4.98 | 1360 | 3294 | 1.68 | 0.0231 | 9.6 |
| 3.75 | 1084 | 1057 | 1.08 | 0.0308 | 12 |
ii. Poly (L-lactide) macromonomer
P(LLA-HEMA) macromonomer was synthesized by the ring opening polymerization by a modified published method [13,82,83]. The FT-IR and 1H-NMR spectra of the synthesized P(LLA-HEMA) macromonomer were consistent with the expected structure [15]. Preliminary synthesis of P(LLA-HEMA) macromonomers with mole fractions of 95 % L-lactide and 5 % HEMA and 90 % L-lactide and 10 % HEMA yielded products of large molecular weights. To reduce molecular weight and improve reactivity, the P(LLA-HEMA) macromonomer with mole fractions of 85 % L-lactide and 15 % HEMA which gave the lowest molecular weight was selected for nanoparticle fabrication. The polydispersity index (PDIpolymer which in polymer chemistry is defined as the ratio of weight average molecular weight (Mw) to the number average molecular weight (Mn) of the polymer or macromonomer) is 1.24, which shows that the macromonomer is monodisperse. The percent composition of HEMA in the macromonomer was determined to be a mole fraction of 3.7 %. The number average molecular weight (Mn) was determined by 1H-NMR (2,085) and GPC (3,420) while the weight average molecular weight (Mw) was determined by GPC (4,247). GPC also revealed a single prominent peak showing that the synthesized macromonomer was pure.
Technique for the Fabrication of Nanoparticles by Dispersion Polymerization [PISA] at Ambient Temperature
Our laboratory has been involved in the application of in-situ dispersion polymerization technique at ambient temperature involving redox initiator system for the fabrication of core-shell nanoparticles (polymerization induces self assembly (PISA) of nanoparticles) [14-19,21,22]. The advantages are similar to those described earlier for in-situ polymerization in the fabrication of nanoparticles. In addition, the uniqueness of in-situ dispersion polymerization at ambient temperature involving organic redox initiator system for the fabrication of core-shell nanoparticles are: nanoparticle fabrication at ambient temperature is suitable for thermolabile bioactive agents like peptide and proteins (especially monoclonal antibodies); it is surfactant free thereby obviating the problems associated with the use of surfactants in injectable liquid preparations; it is a onepot process.
• Fabrication of stealth macromonomer-based PISA nanoparticles by dispersion polymerization
Nanoparticles were fabricated using different amounts of macromonomer (methacrylateterminated poly(lactide) described in 4.2.2.2 above), initiators (BPO-NPDEA redox initiator system (described in Scheme 1 above), hydrolyzable crosslinking agent (N,O-dimethacryolhydroxylamine) and stabilizer (poly(ethylene glycol) n monomethyl ether monomethacrylate (PEGMA)) in a dioxane/DMSO/water solvent system [15]. Smooth, spherical nanoparticles were obtained (Figure 1). An optimized formulation was selected for drug loading and in vivo studies [15] . In vitro release isotherm of paclitaxel from the nanoparticles shows paclitaxel availability for 7 days. In vitro cytotoxicity testing in breast and ovarian cancer cell lines revealed that the nanoparticle formulation, compared with free paclitaxel at the same drug concentration, exhibited similar cytotoxicity for the duration of the study. The cellular uptake of rhodamine-123-loaded nanoparticles shows that the nanoparticles are internalized by MCF-7 breast cancer cells within 1 h. Biodistribution studies have shown that the nanoparticles accumulate in tumor [16] . We carried a similar study on poly(-ɛ-caprolactone) macromonomer as shown in the SEM below (Figure 2) [17].

Figure 1: (A) Scanning electron microscopy image of paclitaxel-loaded stealth PLA-based PISA nanoparticles prepared by insitu dispersion polymerization. (B) TEM image of paclitaxel-loaded nanoparticles prepared by in-situ dispersion polymerization [15,16]

Figure 2: Typical SEM images of blank stealth poly-ε-caprolactone PISA nanoparticles prepared by in-situ dispersion polymerization [5].
• Fabrication of stealth PISA nanoparticles by dispersion polymerization using acidic pH-sensitive crosslinkers
Studies on n-Butyl Acrylate: Our laboratory carried out structure activity studies on acetal crosslinkers capable of responding to the acidic pH environment in tumors (Scheme 3) [19] .

Scheme 3: Synthesis of acetal crosslinkers 1 (di(2-methacryloyloxyethoxy)-[4-methoxyphenyl]methane), 2 (di(2methacryloyloxyethoxy)- [2,4dimethoxyphenyl] methane) and3(di(2-methacryloyloxyethoxy)-[2,4,6trimethoxyphenyl]methane) [18].
We studied the hydrolysis of the three acetal crosslinkers (crosslinkers 1 (di(2-methacryloyloxyethoxy)-[4- methoxyphenyl]methane), 2(di(2-methacryloyloxyethoxy)-[2,4 dimethoxyphenyl]methane) and 3 (di(2- methacryloyloxyethoxy)-[2,4,6 trimethoxyphenyl]methane)). (Figure 3) shows the plot of percentage of acetal crosslinker hydrolyzed versus time in pH 7.4-buffer and pH 5.0-buffer. The rate of hydrolysis of the crosslinkers is faster at pH 5.0 compared to pH 7.4. Moreover, the rate of hydrolysis is fastest for crosslinker 3, followed by crosslinker 2, and crosslinker 1
The difference in the rate of hydrolysis among the crosslinkers can be attributed to the number of methoxy groups present on the benzene ring of the crosslinkers (Scheme 3). It is known that acid catalyzed hydrolyisis of acetals proceeds via protonation of the acetal followed by decomposition of the protonated intermediate to an alcohol and a resonance stabilized carbocation (acetals are readily hydrolyzed back to the aldehyde and corresponding alcohol from which they were formed) [18]. Since crosslinker 3 has three electron donating methoxy groups, it results in the extra stabilization of the carbocation intermediate formed during hydrolysis, which results in an increased rate of hydrolysis[18]. The effect of buffer on the hydrolysis of the crosslinkers (as reported above) is in agreement with the belief that the hydrolysis of acetals is generally first order relative to hydronium ion concentration, making the expected rate of hydrolysis ten times faster with each unit pH decrease [18]. (Figure 3) clearly shows that the rate of hydrolysis is affected by both factors (i.e. type of crosslinker and pH of buffers).

Figure 3: 3D plot showing the effect of the type of crosslinker and buffer on % of crosslinker hydrolyzed at 2 Hours [18]

Scheme 4: Typical scheme for the synthesis of stealth crosslinked poly(n-butyl acrylate) nanoparticles fabricated with acetal crosslinker by free-radical dispersion polymerization technique. Here, the scheme shows the synthesis of blank nanoparticles fabricated with acetal crosslinker 1.
The thrust for the development of pH responsive nanoparticles is to facilitate their response to the acidic cancer environment at the biophase by releasing their encapsulated payload (Figure 4). The pH dependent hydrolysis behavior of the nanoparticles was observed (data not shown here) [18]. It can be attributed to the hydrolysis of the acetal bonds crosslinking poly (n-BA) chains in the nanoparticle bulk structure, as these nanoparticles are designed to undergo expansion in a pH-dependent fashion with maximum release of the encapsulated drug at pH 5, as found within the cellular compartment (endosome), thereby achieving intracellular drug release. Upon cleavage of the acetal, the polymeric nanoparticle changes from a hydrophobic core structure to a hydrophilic one; thus water enters the structure and causes the nanoparticles to degrade and release the contents. The release profiles of docetaxel from the nanoparticles (Figure 5) was found to be very similar to the hydrolysis profiles of the blank nanoparticles. (Figure 5) indicates that the nanoparticles released much more slowly at pH 7.4 over a period of 6 hours as compared to pH 5, where the entire drug was released in less than 4 hours. This similarity in the results of the hydrolysis of blank nanoparticles and in vitro drug availability studies suggests that the release of the docetaxel occurred due to the pH dependent hydrolysis of acetal bonds in the polymeric matrix. Biological work was carried out on the nanoparticles.

Figure 4: Scanning electron micrograph of the docetaxel-loaded nanoparticles synthesized with crosslinker 3

Figure 5: In vitro availability isotherms for docetaxel-loaded nanoparticles fabricated with crosslinker 3 at pH 7.4 and pH 5. (n = 3)
Cytotoxicity studies showed that the nanoparticles were more effective at the same molar amount than the free drug (docetaxel) against cancer cells. Furthermore, LNCaP cells appeared to be the more sensitive to docetaxel than PC3 cells. The cellular uptake studies clearly showed the presence of discrete nanoparticles within the cells in as little as 2 hours [19].
i. Studies on poly-ɛ-caprolactone: We designed and fabricated dual loaded paclitaxel and 17- AAG stealth polymeric nanoparticles using surfactantfree organic redox-initiated dispersion polymerization. The first phase of the work was optimization studies. Aided by a computer software, central composite face-centered (CCF) statistical experimental design in three independent variables (crosslinker, PEG macromonomer, and acid-labile crosslinking agent) and seventeen runs was implemented. Poly-ɛcaprolactone macromonomer and redox-initiator system concentrations were held constant. Nanoparticles were fabricated at ambient temperature. The formation of nanoparticles was confirmed by scanning electron microscopy, which revealed monodispersed, spherical nanoparticles (Figure 6).

Figure 6: Typical scanning electron micrographs of dual-loaded nanoparticles
Seven response variables were evaluated: particle size, paclitaxel drug loading, 17-AAG drug loading, paclitaxel encapsulation efficiency, 17-AAG encapsulation efficiency, in vitro availability of paclitaxel and in vitro availability of 17- AAG (Figure 6) [22].
The second phase involved nanoparticle fabrication and biological studies on the nanoparticles (Figure 7). The combination of factors to give optimal formulation in the first phase are as follows: Crosslinker 0.373 mmoL (di (2-methacryloyloxyethoxy)-[2,4 dimethoxyphenyl] methane (DMDPM; pH sensitive crosslinker)), PEG 1.00981 mmoL (Poly (ethylene glycol) n monomethyl ether mono methacrylate (PEG-MMA, MW=1,000)), stirring speed, 200 rpm, macromonomer 0.28 mmoL and initiator system, 0.594 mmoL). The scanning electron micrographs (morphology of the nanoparticles) show spherical nanoparticles (data no shown). Two breast cancer cell lines (MCF-7 and SKBR-3) were cultured and treated with media only, blank nanoparticles, paclitaxel (as a free drug), 17-AAG (free drug), paclitaxel + 17-AAG combination (as free drugs), and paclitaxel + 17-AAG combination loaded in poly-ɛ-caprolactone stealth nanoparticles. Each drug in the combination was half the concentration of the single free drug. The cytotoxic effects of the paclitaxel treatment and that of the combination (free drug) were found to be similar in both SKBR3 and MCF7 cell lines. Similar cytotoxic effects were observed for the drug combination both in the drug loaded nanoparticles formulation and in the free drug form for both cell lines. Both paclitaxel and 17-AAG were effectively loaded and released from the polymeric nanoparticles. Paclitaxel (free drug), paclitaxel-17AAG combination (free drug), and dual drug-loaded nanoparticles had similar cytotoxic effects on both cell lines. Paclitaxel and 17-AAG combination resulted in synergistic effect: paclitaxel in the combination with 17-AAG was half its original concentration and yielded similar cytotoxic effect. The dose of paclitaxel was reduced without lowering its therapeutic efficacy (Figure 8).

Figure 7: Influence of PEG, stirring speed and crosslinker on 17-AAG release time [h]: (Four dimensional plot)

Figure 8: 3D plot showing the effect of type of treatment after 96 hours (PTX=Paclitaxel solution, 17AAG =17AAG solution and PTX +17AAG=combination drug solution at half concentration of each drug and DLNP=combination drug loaded nanoparticles containing half the concentration of each drug: SKBR3 HER2-positive cancer cell line after).
Applications of Quality By Design (QbD) in the fabrication of coreshell nanoparticles (PISA of nano-objects) in our laboratory
One mission of the drug product development scientist is to develop drug delivery systems that enhance the optimal performance of the bioactive agents. Quality by design (QbD) and process analytical technology (PAT) in pharmaceutical dosage form design and development—already incorporated into automakers’ production principles—involve developing drug formulations and manufacturing processes which ensure predefined drug product specifications. Product and process understanding is a key element of QbD-PAT [5,16,84]. An important aspect of QbD-PAT is to understand how process and formulation variables affect product characteristics, followed by subsequent optimization of these variables vis-à-vis the final specifications. Traditionally, common approaches to this endeavor include varying the levels of one factor or variable at a time and examining the effect on desired parameters (particle size, yield, internalization into cells, efficacy, etc.) One-factor-at-a-time experiments could not reveal interaction of factors, and this approach can be resource intensive in relation to the information generated. It would take forever to optimize nanoparticle property using one-factor-at-a-time experiments because every time you change a factor or its level, you would have to re-optimize all of the other factors. However, design of experiment (DoE) examines all of the variables simultaneously; it enables the investigator to identify the optimum values for the factors much more quickly. At the same time, information that gives a better understanding of how the factors interact can be easily captured. Thus, DoE is a well-established method for identifying important parameters in pharmaceutical dosage form design and for optimizing the parameters with respect to certain specifications [5,15,84-88]. The main approaches to the DoE to be able to examine all of the variables simultaneously are factorial and mixture experimental designs [5, 15].
• D-optimal mixture design for the fabrication of stealth poly-L-lactidebased nanoparticles:
The statistical experimental designs involving mixture methodology is an efficient method for studying products made from components at various levels. In our laboratory we used D-optimal mixture design for experimental design, analysis, and optimization. When a formulation is a mixture of various components (proportion of the constituents) as studied in our work (dispersion polymerization for the fabrication of nanoparticles) and the levels of the components are constrained, D-optimal mixture design is more useful than a factorial design because it accounts for the dependence of response on proportionality of constituents [5,15,17,87]. Scheffe polynomial models were generated to predict particle size (nm) and percent yield for poly-L-lactide-based nanoparticles as functions of the composition of the formulations (20 formulations Table 2). Aided by a computer software simultaneous numerical and graphical optimizations of nanoparticle size and percent yield for poly-L-lactide-based nanoparticles were carried out using the Scheffe polynomial models generated to predict particle size (nm) and percent yield for poly-L-lactide-based nanoparticles as functions of the composition of the formulations [87]. The corresponding model graphs are shown in (Figures 9, 10 and 11).
TABLE 2. Composition and response of D-optimal mixture experimental design for the fabrication of stealth poly-L- lactide-based nanoparticles
| Standard Order | Run order | A : Cross linking agent (mmol) | B: Initiator System (mmol) | C: Stabilizer (PEG-MMA)(mmol) | Response 1 (Particle size : nm) | Response 1 (Percent Yield : %) |
|---|---|---|---|---|---|---|
| 9 | 1 | 0.048 | 0.359 | 0.304 | 297.6 | 56.87 |
| 8 | 2 | 0.056 | 0.285 | 0.215 | 306.6 | 28.99 |
| 1 | 3 | 0.087 | 0.377 | 0.091 | 261.5 | 62.39 |
| 4 | 4 | 0.018 | 0.565 | 0.214 | 270.6 | 35.26 |
| 19 | 5 | 0.087 | 0.377 | 0.091 | 268.1 | 58.82 |
| 12 | 6 | 0.042 | 0.452 | 0.259 | 268.4 | 31.88 |
| 20 | 7 | 0.018 | 0.565 | 0.214 | 295.8 | 61.08 |
| 17 | 8 | 0.087 | 0.183 | 0.304 | 326.4 | 31.13 |
| 14 | 9 | 0.018 | 0.312 | 0.304 | 290.3 | 41.19 |
| 15 | 10 | 0.053 | 0.415 | 0.148 | 322.9 | 45.15 |
| 3 | 11 | 0.055 | 0.625 | 0.115 | 293.1 | 47.87 |
| 4 | 12 | 0.087 | 0.183 | 0.304 | 322.9 | 34.18 |
| 13 | 13 | 0.036 | 0.522 | 0.163 | 320.7 | 43.08 |
| 2 | 14 | 0.018 | 0.466 | 0.091 | 313.1 | 24.43 |
| 11 | 15 | 0.087 | 0.5 | 0.091 | 273.3 | 43.32 |
| 16 | 16 | 0.018 | 0.446 | 0.091 | 293.8 | 35.08 |
| 18 | 17 | 0.055 | 0.625 | 0.115 | 305.9 | 39.22 |
| 6 | 18 | 0.018 | 0.234 | 0.304 | 320.9 | 35.98 |
| 7 | 19 | 0.018 | 0.625 | 0.091 | 295.6 | 44.71 |
| 10 | 20 | 0.087 | 0.394 | 0.198 | 314.9 | 37.59 |

Figure 9: Model graph showing the design space and variation in particle size as a function of the mixture composition. A=crosslinking agent; B=initiators; C=stabilizer; D=macromonomer (polylactide-based nanoparticles)

Figure 10: Model graph showing the design space and variation in percent yield as a function of the mixture composition. A=Crosslinking agent; B=Initiators; C=Stabilizer and D=macromonomer (polylactide-based nanoparticles)

Figure 11: Simultaneous graphical optimization (overlay plot) of the design space variation in particle size and % yield as functions of the mixture composition. A=crosslinking agent; B=initiators; C=stabilizer and D= macromonomer
Following simultaneous numerical optimization of nanoparticle size and percent yield of poly-L-lactidebased nanoparticles using Scheffe polynomial models from which (Figures 9, 10and 11) were generated, four solutions were returned.
Three of the solutions were used to fabricate nanoparticles to compare the predicted values with the actual laboratory values. The observations from the confirmation experiments are within the confirmation 95% prediction interval (95% PI low and 95% PI high), where PI is point prediction showing the confirmation of the models. A typical overlay plot is shown in (Figure 11), where the grey area in the figure is the design space. The focus on particle size and yield in this aspect of the work is based on the fact that particle size plays a key role in determining body distribution of nanoparticles after in vivo administration by injection, and in facilitating their access to cancer cells (internalization) either by passive or active targeting to tumors. Optimization of the nanoparticle fabrication for a high percent yield will make the drug development effort an economic proposition. A similar optimization was done for stealth polycaprolactone nanoparticles[5].
Central composite face-centered design computer optimization of stealth biodegradable polymeric dual-loaded nanoparticles for cancer therapy:
An acidic pH-sensitive acetal crosslinker (di(2- methacryloyloxyethoxy)-[2,4- dimethoxyphenyl]methane) and a poly(ɛ)caprolactone macromonomer were synthesized and characterized [18]. The experimental design used was the response surface method (RSM) which is an efficient way of planning and optimizing experiments; it involves the principles of design of experiments (DOE). DOE is a statistical technique that increases the productivity of the experiments by minimizing the number of experiments involving multiple variables and maximizing the accuracy of results (an advantage of DOE is that it allows for the maximum amount of information to be extracted using the minimum number of experiments). The important property of DOE is that while several factors are varied simultaneously, each factor may be evaluated independently. Box and his co-workers have been quoted as saying that “if the factors do act additively, the DOE design does the job with much more precision than the one-factor-at-a-time method and if the factors do not active additively, DOE, unlike the one factor-at-atime design, can detect and estimate interactions that measure this nonadditivity” [88,89] . We used the central composite face-centered design (CCF) in three independent factors and seventeen runs. Nanoparticles were fabricated by dispersion polymerization technique. Response variables evaluated were: particle size,drug loading, encapsulation efficiency, and in vitro availability
In an optimization problem, the response surface method (RSM) or response surface modelling (RSM) design is often used as described earlier for D-optimal mixture design, which is different from a statistical experimental design whose objective is screening. There are several classical RSM design families. We selected the central composite face-centered design (CCF) in three independent factors and seventeen runs. A DoE approach was used to systematically investigate the effects of PEG concentration, stirrer speed, and crosslinker concentration. A total of 17 different nanoparticle fabrication experiments (including three replicates of the center points to provide an estimate of replicate error: the replicated experiments enable the performance of a lack of fit test) were carried out based on the CCF design. The factors and data are shown in (Tables 3 and 4) respectively. Macromonomer and the initiator system were held constant, making the experimental design to be a central composite face-centered design (CCF) in three independent factors and seventeen runs. The response variables are nanoparticle size, paclitaxel drug loading, 17-AAG drug loading, paclitaxel encapsulation efficiency, 17-AAG encapsulation efficiency, release time for paclitaxel and release time for 17-AAG. All experiments were performed in a random order to minimize the effects of uncontrolled factors that might introduce a bias into the measurements (Tables 3 and 4)
TABLE 3. Values of factors used in the fabrication of nanoparticles
| Experiment No | Run order | Cross Linker (mmol) | PEG (mmol) | Stirring speed(rpm) | Macromonomer (mmol) | Initiator system(mmol) |
|---|---|---|---|---|---|---|
| 1 | 6 | 0.373 | 0.898 | 100 | 0.28 | 0.594 |
| 2 | 17 | 0.466 | 0.898 | 100 | 0.28 | 0.594 |
| 3 | 12 | 0.373 | 1.123 | 100 | 0.28 | 0.594 |
| 4 | 11 | 0.466 | 1.123 | 100 | 0.28 | 0.594 |
| 5 | 16 | 0.373 | 0.898 | 300 | 0.28 | 0.594 |
| 6 | 9 | 0.466 | 0.898 | 300 | 0.28 | 0.594 |
| 7 | 15 | 0.373 | 1.123 | 300 | 0.28 | 0.594 |
| 8 | 7 | 0.466 | 1.123 | 300 | 0.28 | 0.594 |
| 9 | 8 | 0.373 | 1.0105 | 200 | 0.28 | 0.594 |
| 10 | 13 | 0.466 | 1.0105 | 200 | 0.28 | 0.594 |
| 11 | 4 | 0.4195 | 0.898 | 200 | 0.28 | 0.594 |
| 12 | 3 | 0.4195 | 1.123 | 200 | 0.28 | 0.594 |
| 13 | 10 | 0.4195 | 1.0105 | 100 | 0.28 | 0.594 |
| 14 | 1 | 0.4195 | 1.0105 | 300 | 0.28 | 0.594 |
| 15 | 2 | 0.4195 | 1.0105 | 200 | 0.28 | 0.594 |
| 16 | 5 | 0.4195 | 1.0105 | 200 | 0.28 | 0.594 |
| 17 | 14 | 0.4195 | 1.0105 | 200 | 0.28 | 0.594 |
TABLE 4. Data on nanoparticle properties (Response variables to the factors shown in Table 3, where P=paclitaxel, and G=17AAG; ε = standard deviation
| Exp. No | Run order | Particle Size (nm) | Drug Loading P(%) | Drug Loading G(%) | Encapsulation Efficiency (Paclitaxel)(%) | Encapsulation Efficiency (17-AAG)(%) | Release Time Drug (Paclitaxel) (h) | Release Time Drug (17-AAG) (h) |
|---|---|---|---|---|---|---|---|---|
| 1 | 6 | 291.7 | 1.53 | 0.83 | 98.76 | 97.99 | 50.7 | 24.3 |
| 2 | 17 | 298.4 | 1.62 | 0.89 | 98.47 | 97.61 | 48 | 48 |
| 3 | 12 | 267.5 | 1.64 | 0.88 | 90 | 90.49 | 60.8 | 24 |
| 4 | 11 | 270.6 | 1.76 | 0.95 | 99.75 | 98.8 | 74 | 59 |
| 5 | 16 | 228.6 | 1.58 | 0.88 | 99.75 | 99.37 | 49 | 49 |
| 6 | 9 | 261.2 | 1.53 | 0.85 | 95.85 | 93.77 | 49 | 25 |
| 7 | 15 | 217.1 | 1.99 | 0.98 | 99.87 | 98.93 | 48 | 48 |
| 8 | 7 | 260.8 | 1.79 | 0.93 | 99.93 | 98.98 | 72 | 72 |
| 9 | 8 | 229 | 1.57 | 0.95 | 92.45 | 91.75 | 48 | 24 |
| 10 | 13 | 289.6 | 1.71 | 0.85 | 97.3 | 95.25 | 48 | 32 |
| 11 | 4 | 286.1 | 1.72 | 0.9 | 89.78 | 91.5 | 72 | 36 |
| 12 | 3 | 255.3 | 1.94 | 0.86 | 97.91 | 95.68 | 68 | 48 |
| 13 | 10 | 250.3 | 1.85 | 0.97 | 99.22 | 97.63 | 71 | 24 |
| 14 | 1 | 288.7 | 1.65 | 0.92 | 97.02 | 97.58 | 74 | 48.3 |
| 15 | 2 | 249.4 | 1.78 | 1 | 94.47 | 96.88 | 70 | 28 |
| 16 | 5 | 235.6 | 1.87 | 0.96 | 96.98 | 97.75 | 73 | 24 |
| 17 | 14 | 232.4 | 1.87 | 1 | 98.69 | 97.04 | 69 | 22 |
| € | ± 0.2- ± 10.6 | ±0.02- ± 0.015 | ±0.01- ± 0.003 | ± 0.029 - ± 0.002 | ± 0.450 - ± 0.118 | ± 0.31 - ± 0.037 | ± 0.029 - ± 0.046 |
Aided by a computer software statistical analysis of the data was performed. A quadratic model was fitted to the data using multiple linear regression to be able to determine the regression coefficients. The significance of the model was tested by the analysis of variance (ANOVA) with a 95% confidence level. The quadratic model for each response variable will have 10 terms: one constant, three linear, three quadratics, and three two-factor interactions (Equation 1)
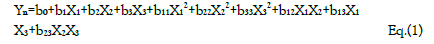
Yn is the dependent variable; b0 is the model constant; b1 , b2 , and b3 show the effects of corresponding or related variable on the response variables; b11 , b22 and b22 are the quadratic coefficients and b12, b13, and b23 are the interaction coefficients between the studied factors.
Using the data obtained from all the analyses, the optimization objectives were set and an optimized formulation was generated using the optimizer option in the software following specifications for each of the response variables (Table 5) with predicted outcomes for particle size, drug loading, encapsulation efficiency, and release time. The run with the lowest log D was selected (Log D = -1.46 with the probability of failure of 2.2%). Log D value is known to indicate the weighted average of the individual response desirabilities. The lower the value of Log D, the better the optimization. From (Table 5), crosslinker has the highest contribution (44.80%) to the optimized formulation followed by stirring speed (35.02%) and then PEG (20.15%). The predicted and actual responses for the formulation generated by the optimizer are displayed in (Table 6), showing that the actual response is within the 95% confidence intervals

TABLE 5: Results of optimization of the data
TABLE 6. Predicted and actual responses of optimized formulation
| Response variable | Predicted Response and 95% Confidence Intervals | Actual Response | ||
|---|---|---|---|---|
| Lower Bowndary | Upper Boundary | |||
| Particle size | 244.0 nm | 225.4 nm | 259.8nm | 243.6 nm ± 0.50 |
| Drug Loading P | 1.66% | 1.59% | 1.74% | 1.71% ± 0.03 |
| Drug Loading G | 0.96% | 0.90% | 1.02% | 0.92% ± 0.06 |
| Encapsulation Efficiency P | 96.99% | 91.08% | 98.99% | 93.03% ± 0.4 |
| Encapsulation Efficiency G | 96.12% | 93.45% | 97.70% | 97.83% ± 0.01 |
| Release time P | 50.92 h | 46.22 h | 46.22 h | 53.11h ± 0.07 |
| Release time G | 24.29 h | 15.6 h | 15.61 h | 30h ± 0.03 |
Conclusion
Polymerization Induced Self-Assembly (PISA) for the fabrication of nanoparticles is believed to be a better strategy than nanoparticle fabrication from preformed polymers because it is very difficult to tether targeting ligands, like mAbs, to the corona/surface of the nanoparticles for biorecognition events. Any attempt to modify the surface of nanoparticles fabricated by dispersion of preformed polymers often results in a substantial loss of encapsulated bioactive agents, contrast agents for imaging or other materials. Further, generally, creation of nanostructures via self-assembly of preformed block copolymers is performed in low concentrations and involves multiple steps which prevent its commercialization and further applications. The preparation of multi-morphologies via polymerization-induced self-assembly and morphology transition can be conducted with the monomer concentration as high as 500 mg mL-1 which is not possible with selfassembly of preformed block copolymers in a selective solvent, in which 1 mg mL-1 of copolymer is often used. Among the techniques available for in situ polymerization for the fabrication of nanoparticles are emulsion polymerization, microemulsion polymerization, miniemulsion polymerization, dispersion polymerization, and suspension polymerization. The dispersion polymerization has been shown to be the best. RDRP processes such as Atom Transfer Radical Polymerization (ATRP), Reversible Addition Fragmentation Chain Transfer (RAFT) polymerization and nitroxide mediated polymerization (NMP) have revolutionized polymer synthesis by providing polymer chemists with powerful tools that enable control over architecture, composition, molecular weight and chain length distributions. RDRP processes, especially RAFT polymerization, are embraced in PISA. The technique for the fabrication of nanoparticles by dispersion polymerization (PISA) at an ambient temperature was described with examples from our laboratory involving organic redox initiated polymerization using the FDA approved biodegradable polymers. Response surface methodology and computer optimization are useful in the understanding the factors that ensure the optimized properties of drug-loaded nanoparticles
Acknowledgment And Funding
he author acknowledges with thanks funding support from NIH/NIGMS: 1R16GM145483-01. This work was carried out in facilities supported by NCRR/NIH Grants #1C06 RR 020608-01 and #1 C06 RR 14469-01.
Conflicts of Interest
The author declares no conflict of interest
Declaration of Interests
The author declares that he has no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Akala EO, Adesina S. Fabrication of Polymeric Core-Shell Nanostructures. In: Alexandru Mihai Grumezescu., ed. Nanoscale Fabrication, Optimization, Scale-up and Biological Aspects of Pharmaceutical Nanotechnology. Applied Science Publishers: 2017:1-49.
- Krukemeyer MG, Krenn V, Huebner F, Wagner W, Resch R. History and possible uses of nanomedicine based on nanoparticles and nanotechnological progress. J. Nanomed. Nanotechnol. 2015 Nov 1;6(6):336.
- Akala EO. Strategies for transmembrane passage of polymer-based nanostructures In: Broz, P, ed. Polymer-based nanostructures: Medical applications. RSC Nanosci Nanotech. Royal Soc Chem, 2010;16-80.
[GoogleScholar] [CrossRef]
- Hillaireau H, Couvreur P. Polymeric nanoparticles as drug carriers. Polym. drug deliv. 2006 May 19:101-10.
- Ogunwuyi O, Adesina S, Akala EO. d-Optimal mixture experimental design for stealth biodegradable crosslinked docetaxel-loaded poly-ε-caprolactone nanoparticles manufactured by dispersion polymerization. Die Pharmazie-An Int J Pharmaceutical Sci. 2015 Mar 2;70(3):165-76.
- Kelkar SS, Reineke TM. Theranostics: combining imaging and therapy. Bioconjugate chemistry. 2011 Oct 19;22(10):1879-903.
- Bhaskar S, Tian F, Stoeger T, et al. Multifunctional Nanocarriers for diagnostics, drug delivery and targeted treatment across blood-brain barrier: perspectives on tracking and neuroimaging. Particle and fibre toxicology. 2010;7(1):1-25.
- Drbohlavova J, Chomoucka J, Adam V, et al. Nanocarriers for anticancer drugs-new trends in nanomedicine. Current Drug Metabo. 2013;14(5):547-64.
[GoogleScholar] [CrossRef]
- Su H, Wang Y, Gu Y, et al. Potential applications and human biosafety of nanomaterials used in nanomedicine. J Applied Toxico 2018;38(1):3-24.
- Davis ME. Z. Chen and DM Shin. Nat. Rev. Drug Discovery. 2008;7:771-82.
- Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS nano. 2013 Nov 26;7(11):9518-25.
- Hu CM, Zhang L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem Pharmaco. 2012;83(8):1104-1111.
- Sparreboom A, Baker SD, Verweij J. Paclitaxel repackaged in an albumin-stabilized nanoparticle: handy or just a dandy? J Clinical Onco. 2005;23(31):7765-7767.
- Akala EO, Okunola O. Novel stealth degradable nanoparticles prepared by dispersion polymerization for the delivery of bioactive agents Part I. Pharm Ind. 2013;75(7):1191-1196.
- Adesina SK, Wight SA, Akala EO. Optimization of the fabrication of novel stealth PLA-based nanoparticles by dispersion polymerization using D-optimal mixture design. Drug Dev Industrial Pharm. 2014;40(11):1547-1556.
- Adesina SK, Holly A, Kramer-Marek G, et al. Polylactide-based paclitaxel-loaded nanoparticles fabricated by dispersion polymerization: characterization, evaluation in cancer cell lines, and preliminary biodistribution studies. J Pharmaceutical Sci. 2014;103(8):2546-2555.
- Ogunwuyi O, Adesina S, Akala EO. d-Optimal mixture experimental design for stealth biodegradable crosslinked docetaxel-loaded poly-ε-caprolactone nanoparticles manufactured by dispersion polymerization. Die Pharmazie-An Int J Pharmaceutical Sci. 2015;70(3):165-176.
- Ogunwuyi O, Kumari N, Smith KA, et al. Antiretroviral drugs-loaded nanoparticles fabricated by dispersion polymerization with potential for HIV/AIDS treatment. Infectious Dis: Res Treatm. 2016.
- Puri R, A Berhe S, O Akala E. pH-sensitive polymeric nanoparticles fabricated by dispersion polymerization for the delivery of bioactive agents. Pharm Nanotechno. 2017;5(1):44-66.
- Puri R, Adesina S, Akala E. Cellular uptake and cytotoxicity studies of pH-responsive polymeric nanoparticles fabricated by dispersion polymerization. J Nanosci Nanomed 2018;2(1):3.
- Fisusi F, Brandy N, Wu J, et al. Studies on polyethylene glycol-monoclonal antibody conjugates for fabrication of nanoparticles for biomedical applications. J Nanosci Nanomed. 2020;4(2):1.
- Berko YA, Funmilola AF, Akala EO. Fabrication of Paclitaxel and 17AAG-loaded Poly-ε-Caprolactone Nanoparticles for Breast Cancer Treatment. J Pharmace Drug Delivery Res. 2021;10(1).
- Berko YA, Akala EO. Computer Optimization of Stealth Biodegradable Polymeric Dual-loaded Nanoparticles for Cancer Therapy Using Central Composite Face-centered Design. Pharma Nanotechn. 2020;8(2):108-32.
- Lammers T, Kiessling F, Henning WE., et al. Nanotheranostics and image guided drug delivery: current concepts and future directions. Mol Pharmace 2010;7 (6):1899-1912.
- Ahmed N, Fessi H, Elaissari A. Theranostic applications of nanoparticles in cancer. Drug discovery today. 2012;17(17-18):928-934.
- Artemov D, Mori N, Okollie B, et al. MR molecular imaging of the Her‐2/neu receptor in breast cancer cells using targeted iron oxide nanoparticles. Magnetic Res Med: An Official J Int Soc Mag Reson Med. 2003 Mar;49(3):403-408.
- Bogin L, Margalit R, Mispelter J, et al. Parametric imaging of tumor perfusion using flow‐and permeability‐limited tracers. J Magn Reson Imaging: Off J Int Soc Magn Reson Med. 2002;16(3):289-299.
- Glunde K, Jacobs MA, Pathak AP, et al. Molecular and functional imaging of breast cancer. NMR Biomed.: Int J Devoted Devel App Magn Reson Vivo 2009 Jan;22(1):92-103.
- Wan WM, Hong CY, Pan CY, et al. One-pot synthesis of nanomaterials via RAFT polymerization induced self-assembly and morphology transition. Chem Commun., 2009(39):5883-5885.
- Landfester K, Mailänder V. Nanocapsules with specific targeting and release properties using miniemulsion polymerization. Expert opin drug deliv. 2013;10(5):593-609.
- Kelkar SS, Reineke TM. Theranostics: combining imaging and therapy. Bioconjugate chem. 2011;22(10):1879-1903.
- Arshady R. Suspension, emulsion, and dispersion polymerization: A methodological survey. Colloid polym sci. 1992 ;270(8):717-32.
- Landfester K, Musyanovych A, Mailänder V. From polymeric particles to multifunctional nanocapsules for biomedical applications using the miniemulsion process. J Polym Sci A: Polym Chem.2010;48(3):493-515.
- Song JS, Tronc F, Winnik MA. Monodisperse, controlled micron-size dye-labeled polystyrene particles by two-stage dispersion polymerization. Polymer. 2006;47(3):817-825.
- Ha ST, Park OO, Im SH. Size control of highly monodisperse polystyrene particles by modified dispersion polymerization. Macromol Res. 2010;18(10):935-9543.
- Han H, Lee J, Hong J, et al. Dispersion polymerization of styrene employing lyophilic comonomer in the absence of stabilizer: synthesis of impurity-free microspheres. Macromol Res. 2009;17(7):469-475.
- Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Delivery. 2013.
- Boyer C, Bulmus V, Davis TP, et al. Bioapplications of RAFT polymerization. Chemical Rev. 2009;109(11):5402-5436.
- Corrigan N, Jung K, Moad G, et al. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Progr Polymer Sci. 2020; 111:101311.
- Pearce S, Perez-Mercader J. PISA: construction of self-organized and self-assembled functional vesicular structures. Polymer Chem. 2021;12(1):29-49.
- Farokhzad OC, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv. drug deliv. rev. 2006;58(14):1456-9.
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005 Mar;5(3):161-171.
- Lansalot M, Rieger J. Polymerization‐Induced Self‐Assembly. Macromole Rapid Commun.2019 ;40(2):1800885.
- Perrier S. 50th Anniversary Perspective: RAFT Polymerization A User Guide. Macromolecules. 2017;50(19):7433-7447.
- Cornel EJ, Jiang J, Chen S, et al. Principles and characteristics of polymerization-induced self-assembly with various polymerization techniques. CCS Chemistry. 2021;3(4):2104-25.
- Derry MJ, Fielding LA, Armes SP. Industrially-relevant polymerization-induced self-assembly formulations in non-polar solvents: RAFT dispersion polymerization of benzyl methacrylate. Polym Chem. 2015;6(16):3054-3062.
- Blanazs A, Ryan AJ, Armes SP. Predictive phase diagrams for RAFT aqueous dispersion polymerization: effect of block copolymer composition, molecular weight, and copolymer concentration. Macromolecules. 2012;45(12):5099-5107.
- Warren NJ, Derry MJ, Mykhaylyk OO, et al. Critical dependence of molecular weight on thermoresponsive behavior of diblock copolymer worm gels in aqueous solution. Macromolecules. 2018;51(21):8357-8371.
- Li Y, Armes SP. RAFT synthesis of sterically stabilized methacrylic nanolatexes and vesicles by aqueous dispersion polymerization. Angew Chem. 2010;122(24):4136-40.
- Fielding LA, Derry MJ, Ladmiral V, et al. RAFT dispersion polymerization in non-polar solvents: facile production of block copolymer spheres, worms and vesicles in n-alkanes. Chem Sci. 2013;4(5):2081-7.
- Blanazs A, Madsen J, Battaglia G, et al. Mechanistic insights for block copolymer morphologies: how do worms form vesicles?. J Am Chem Soc. 2011;133(41):16581-7.
- Canton I, Warren NJ, Chahal A, et al. Mucin-inspired thermoresponsive synthetic hydrogels induce stasis in human pluripotent stem cells and human embryos. ACS cent Sci. 2016;2(2):65-74.
- Cao C, Chen F, Garvey CJ, et al. Drug-directed morphology changes in polymerization-induced Self-assembly (PISA) influence the biological behavior of nanoparticles. ACS Appl Mat Interfaces.2020 Jun 9;12(27):30221-30233..
- Chen M, Zhong M, Johnson JA. Light-controlled radical polymerization: mechanisms, methods, and applications. Chem Rev. 2016;116(17):10167-211.
- Yeow J, Sugita OR, Boyer C. Visible light-mediated polymerization-induced self-assembly in the absence of external catalyst or initiator. ACS Macro Letters. 2016 May 17;5(5):558-564.
- Sun H, Cao W, Zang N, et al. Proapoptotic Peptide Brush Polymer Nanoparticles via Photoinitiated Polymerization‐Induced Self‐Assembly. Angew Chem. 2020;132(43):19298-304.
- Tan J, Sun H, Yu M, et al. Photo-PISA: shedding light on polymerization-induced self-assembly. ACS Macro Letters. 2015;4(11):1249-1253.
- Jiang Y, Xu N, Han J, et al. The direct synthesis of interface-decorated reactive block copolymer nanoparticles via polymerisation-induced self-assembly. Polymer Chem. 2015;6(27):4955-4965.
- Wang G, Schmitt M, Wang Z, et al. Polymerization-induced self-assembly (PISA) using ICAR ATRP at low catalyst concentration. Macromolecules. 2016 Nov 22;49(22):8605-8615.
- Alzahrani A, Zhou D, Kuchel RP, et al. Polymerization-induced self-assembly based on ATRP in supercritical carbon dioxide. Polym Chem. 2019;10(21):2658-2665.
- Moad G, Rizzardo E, Thang S, et al. Toward living radical polymerization. Acc chem res. 2008 Sep 16;41(9):1133-42.
- Audran G, Bagryanskaya EG, Marque SR, et al. New variants of nitroxide mediated polymerization. Polymers. 2020 Jul 2;12(7):1481.
- Delaittre G, Nicolas J, Lefay C, et al. Aqueous Suspension of Amphiphilic Diblock Copolymer Nanoparticles Prepared In Situ from a Water-Soluble Poly (Sodium Acrylate) Alkoxyamine Macroinitiator. Soft Matter, 2006;2:223–231.
- Delaittre G, Save M, Charleux B. Nitroxide-Mediated Aqueous Dispersion Polymerization: From Water-Soluble Macroalkoxyamine to Thermosensitive Nanogels. Macromol. Rapid Commun. 2007;28:1528–1533.
- Delaittre G, Dire C, Rieger J, et al. Formation of Polymer Vesicles by Simultaneous Chain Growth and Self-Assembly of Amphiphilic Block Copolymers, Chem. Commun. 2009;2887–2889.
- Sarac AS. 1999. Redox polymerization. Progre Polymer Sci 24;1149-1204.
- Odian G, Principles of polymerization, 2004. Wiley-Intersci., New York.
- Blanca V, Levenfeld B, Roman JS. 1998. Role of amine activators on the curing parameters, properties and toxicity of acrylic bone cements. Polymer Int.1998; 46: 241-250.
- Vazquez B, Belen L, Julio SR, et al. Role of amine activators on the curing parameters, properties and toxicity of acrylic bone cements. Polym Int. 1998;46: 241-250.
- Paul DR. Controlled release polymeric formulations. ACS symposium series 33. Am Chem Soc. Washington, DC, 1976;1-14.
- Malinova V, Wolfgang Meier W. Polymer materials for biomedical applications. In: Broz P. ed., Polymer-based nanostructures: Medical applications. RSC nanoscience and nanotechnology, series 9. R Soc Chem. Cambridge, UK, 2010;3-15.
- Akala EO. Strategies for transmembrane passage of polymer-based nanostructures. In: Broz P. ed. Polymer-based nanostructures: Medical applications. RSC nanoscience and nanotechnology, series 9. R Soc Chem. 2010;16-80.
- Schindler A, Jeffcoat R. Kimmel GL, et al. Biodegradable polymers for sustained drug delivery. Contemp Top Polym Sci., 1977:2:251-289.
- Pitt GG, Gratzl MM, Kimmel GL, et al. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (ε-caprolactone), and their copolymers in vivo. Biomaterials. 1981 Oct 1;2(4):215-20.
- Woodward SC, Brewer PS, Moatamed F, et al. The intracellular degradation of poly(epsilon caprolactone). J Biomed Mater Res. 11985;9: 437-444.
- Liu Y, Schulze M, Albertsson AC. α-Methacryloyl-ω-hydroxylpoly(ɛ-caprolactone) macromonomer: synthesis, characterization, and copolymerization J Macromol Sci Pure Appl Chem, 1998;35: 207–232.
[GoogleScholar] [CrossRef]
- Tortosa K, Miola C, Hamaide T. Synthesis of low molecular weight-hydroxy polycaprolactone macromonomers by coordinated anionic polymerization in protic conditions J Appl Polym Sci, 1997; 65:2357–2372.
- Akala EO, Wiriyacoonkasem P, Pan G. Studies on in vitro availability, degradation, and thermal properties of naltrexone-loaded biodegradable microspheres. Drug Dev Ind Pharm 2011;37: 673–684.
- Dubois PH, Jerome R, Teyssle PH. Macromolecular engineering of polylactones and polylactides. 3. synthesis, characterization, and applications of poly(e-caprolactone) macromonomers. Macromolecules, 1991; 24: 977–981.
- Czelusniak I, Khosravi E, Kenwright AM, et al.. Synthesis, characterization and hydrolytic degradation of polylactide-functionalized polyoxanorbornenes. Macromolecules, 2007;40: 1444–1452.
- Ryner M, Finne A, Albertsson Ac, et al.. L-lactide Macromonomer synthesis initiated by new cyclic tin alkoxides functionalized for brushlike structures. Macromolecules 2001;34: 7281–7287.
- Huang SJ, Onyari JM. Multicomponent polymers of poly(lactic acid) macromonomers with methacrylate terminal and copolymers of poly(2-hydroxyethyl methacrylate). JMS Pure Appl Chem A. 1996; 33(5):571–584.
- Yu, L.X., 2008. Pharmaceutical quality by design: product and process development, understanding, and control. Pharmaceutical Research 10, 781-791.
- Yu LX. Gregory A, Khan MA, Hoag, SW, et al. Understanding pharmaceutical quality by design AAPS J. 2014;16(4):771-83.
- Yu, L.X. and J. Woodcock. FDA pharmaceutical quality oversight. Int J Pharm. 491 (2015) 2–7.
- Yu LX, Amidon G, Khan MA, et al . Understanding pharmaceutical quality by design. The AAPS journal. 2014 Jul;16(4):771-83. [Google Scholar] [CrossRef]
- Lawrence XY, Woodcock J. FDA pharmaceutical quality oversight. Int j pharm. 2015 Aug 1;491(1-2):2-7. [Google Scholar] [CrossRef]
- Akala EO, Adesina S, Ogunwuyi O, et al. Computer optimization of biodegradable nanoparticles fabricated by dispersion polymerization. Int J Environ Res Public Health. 2016 Jan;13(1):47.
- Fenech A, Fearn T, Strlic M. Use of Design-of-Experiment Principles to develop a dose-response function for color photographs. Polym Degrad Stabil 2012; 97: 621-5.
- George EP, Hunter WG, Hunter JS, et al. Statistics for experimenters: design, innovation, and discovery. Wiley; 2005 May.