The role of the sympathetic nervous system in sudden cardiac death
- *Corresponding Author:
- Dr Naranjan S Dhalla
Institute of Cardiovascular Sciences, St Boniface Research Centre, 351 Tache Avenue, Winnipeg, Manitoba R2H 2A6
Tel: 204-235-3417, fax 204-237-0347
E-mail: nsdhalla@srbc.ca
This open-access article is distributed under the terms of the Creative Commons Attribution Non-Commercial License (CC BY-NC) (http:// creativecommons.org/licenses/by-nc/4.0/), which permits reuse, distribution and reproduction of the article, provided that the original work is properly cited and the reuse is restricted to noncommercial purposes. For commercial reuse, contact reprints@pulsus.com
[ft_below_content] =>Keywords
Antioxidants; ß-adrenoceptor blockers; Cardiac arrhythmias; Catecholamines; Sudden cardiac death; Sympathetic nervous system
Sudden cardiac death (SCD) is characterized as unforeseen failure of the heart due to cardiac arrhythmias, resulting in acute symptoms including palpitations, vomiting and fatigue, followed by severe symptoms such as rapid syncope, loss of breathing, loss of pulse, sudden collapse and, eventually, death (1,2). Most sudden deaths are related to cardiac issues, accounting for more than 15% to 20% of global deaths (300,000 to 400,000 in the United States, depending on the definition) per year (3,4). More than 50% of these deaths occur as the initial manifestation of an underlying heart disease, with cardiac arrest occurring within 1 h to 2 h of symptom onset, in what would normally be considered a low-risk individual (1,5). The underlying disease may result in either structural defects or electrical abnormalities, both of which can lead to serious ventricular arrhythmias and, eventually, SCD through multiple mechanisms (3). Occurrence of structural, electrical and metabolic changes, as well as electrolyte distribution alterations that can occur due to channelopathies, have been proposed to explain SCD. However, sustained sympathetic activation in response to a stressful stimulus and subsequent release of catecholamines and their oxidation has been suggested to be a unifying mechanism of SCD (1,3). Despite the fact that a large proportion of SCD victims exhibited coronary artery disease (CAD), irregular electrocardiographic patterns and ventricular remodelling due to myocardial infarction or vascular anomaly, risk stratification is challenging because the same pathophysiological indicators are present in many patients who have not experienced SCD (6,7). Aside from drugs modulating ion channels and drugs affecting the renin-angiotensin system, typical therapy of high-risk patients involves the use of drugs (ß-adrenoceptor blockers) that oppose the effects of catecholamines as well as the implantation of cardioverter defibrillators (1,8). Both the pathogenesis and the prevention/treatment of SCD have been extensively reviewed in the recent literature (9-14) and, based on these, the American College of Cardiology (ACC)/American Heart Association (AHA) as well as European Society of Cardiology (ESC) have updated their guidelines regarding this topic. The purpose of the present article is to briefly discuss the underlying etiologies, risk stratification methods and treatment strategies for SCD, and provide a detailed assessment of the mechanisms associated with SCD, mainly the release and subsequent oxidation of excessive catecholamines through the sympathetic stress response.
Etiology
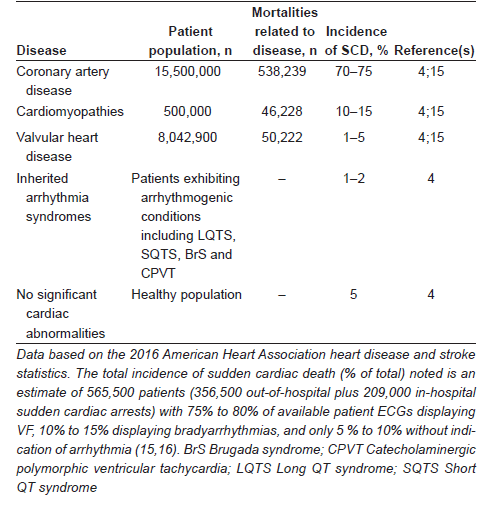
According to the 2016 Heart Disease and Stroke Statistics published by the AHA, the incidence of (emergency medical services assessed) out-of-hospital sudden cardiac arrest is approximately 110.8 per 100,000 individuals (356,500 individuals per year) in the United States, and the incidence of in-hospital sudden cardiac arrest is approximately 209,000 (15). The average survival rate of patients who experience an out-of-hospital sudden cardiac arrest is 10.8% and, for those who experience an in-hospital sudden cardiac arrest is 18.4% (15). The majority of patients who experience SCD are adult, with <1% of SCDs occurring in individuals <35 years of age; the incidence of SCD is higher in men than in women (4). While channelopathies and cardiomyopathies, myocarditis and substance abuse are the major cause of SCD in young subjects, chronic degenerative diseases (CAD, valvular heart diseases and HF) predominate in the pathogenesis of the disease in older populations. Table 1 summarizes the incidence of SCD in the different cohorts indicated based on the most recent sudden cardiac arrest statistics (4,15).
Cardiac arrhythmias are the most common cause of SCD; these involve irregularity of electrical activity, the most common form associated with SCD being ventricular fibrillation (VF), which causes desynchronization of ventricular contractions (1,3). Ventricular arrhythmias are encountered in almost all SCD patients, 75% to 80% exhibiting VF, 10% to 15% exhibiting bradyarrhythmias and only 5% to 10% without indication of arrhythmia (16). This disruption in the order of the transmittance of the excitation wave (generated at the sinoatrial node) results in improper beating of the heart and, thus, incomplete pumping and circulation of blood (17). Arrhythmias in patients without structural disease/irregularities result from primary electrical abnormalities, including Wolff-Parkinson-White (WPW) syndrome, long-QT syndrome (LQTS), short-QT syndrome (SQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT) and bradycardias (3). These observations can form the basis for the use of defibrillators for the correction of arrhythmias and prevention of SCD. Becuase the incidence of SCD is dependent on the type of certain cardiac diseases, it is considered appropriate to discuss this aspect in some detail.
CAD and Cardiomyopathies
The most common cause of SCD is CAD, which develops as a result of damage or disease (typically due to plaque formation and inflammation) affecting the coronary arteries, which supply oxygenated blood and nutrients to the heart (18). CAD is the most common cause of SCD in adults >40 years of age, with atherosclerotic CAD accounting for 75% to 80% of the cases in this cohort (19). It also needs to be emphasized that most of these SCD cases involve seemingly normal adults with no symptoms or history of CAD; therefore, some caution should be exercised for explaining the mechanisms of SCD in patients with CAD (20).
Similar to ischemic cardiomyopathy, both hypertrophic and dilated cardiomyopathies are conditions that involve abnormalities of the heart muscle, diminishing the ability of the heart to pump blood or maintain normal electrical rhythm (21). Cardiomyopathies commonly associated with SCD include hypertrophic cardiomyopathy, arrhythmogenic right ventricular dysplasia, and dilated cardiomyopathy (1). Hypertrophic cardiomyopathy is a heritable autosomal dominant disorder and is the most common cause of SCD in young athletes. It involves abnormal growth of cardiomyocytes, causing thickening of the heart’s muscular walls and cardiomyocyte misalignment (myocardial disarray), causing them to become nonfunctional (1,22).Arrhythmogenic right ventricular dysplasia is another inherited cardiomyopathy that involves predominantly right ventricular fibro-fatty replacement of cardiomyocytes, predisposing patients to lethal arrhythmias associated with SCD (1,23). Finally, dilated cardiomyopathy involves both left ventricular dilation and systolic dysfunction, resulting in cardiac remodelling and, consequently HF, and is the second leading cause of SCD after CAD (24,25).
Valvular disorders
Valvular disorders, including aortic stenosis and mitral valve prolapse, can also manifest ventricular arrhythmias, prompting SCD (1,26). Aortic stenosis is the narrowing of aortic valve outlet of the left ventricle, restricting blood flow to the aorta, causing shortness of breath or breathlessness, angina, palpitations, heart murmurs and syncope (27). If left untreated, a decrease in coronary blood flow and overall decreased cardiac output due to acute left ventricular failure can result in SCD (1). On the other hand, mitral valve prolapse is caused by uneven closure of the mitral valve, causing it to protrude (prolapse) into the left atrium, resulting in the backward leak of small amounts of blood (28). Even in the absence of notable hemodynamic impairment, mitral valve prolapse can produce ventricular arrhythmias and SCD (26).
Diabetes mellitus
Diabetes mellitus comprises a collection of metabolic diseases, which cause hyperglycemia rooted in faults in either insulin secretion due to pancreatic β-cell dysfunction as well as abnormalities resulting in resistance to insulin action, or both, affecting >382 million individuals worldwide (29,30). Acute symptoms of hyperglycemia include polyuria, polydipsia, weight loss and blurred vision (29). Long-term induction of hyperglycemia is linked with the impairment of growth, increased susceptibility to infections, and damage and dysfunction of various organs, mainly the eyes, kidneys, heart and blood vessels, as well as nerves (29). The risk for SCD is significantly increased in diabetes mellitus patients, with large-group studies indicating a two to four times increase in the incidence of SCD compared with the normal population (30). SCD is one of the most prominent causes of death in patients with diabetes, accounting for approximately 50% of deaths due to cardiovascular causes in this cohort (30,31).
Mechanisms of SCD
Several mechanisms have been proposed in an attempt to explain SCD.These include the presence of vascular anomaly and mainly cardiac structural, electrical, metabolic and electrolyte abnormalities, as well as genetic variations (Figure 1). Structural heart disease can lead to the development of SCD, giving rise to the postulation that SCD is caused solely by structural abnormalities in CAD, ischemic cardiomyopathies, congenital heart disease, valvular heart diseases and various cardiomyopathies (3). Electrophysiological/electrical abnormalities have also been considered as a possible mechanism for SCD, accrediting SCD to the arrhythmogenic primary electrical abnormalities including WPW, LQTS, SQTS, BrS and CPVT. The appearance of focal activity, such as enhanced automaticity and triggered activity or reentry, which can occur due to an anatomical or functional obstacle, has also been suggested to result in SCD (3,17). Lifethreatening arrhythmias, which can lead to SCD, are also known to be associated with electrolyte imbalances, such as hypokalemia, but also hyperkalemia and hypomagnesemia, metabolic abnormalities, severe inflammation and endocrine disorders (hyper- and hypothyroidism) (3). More recently, several genetic variations, including those affecting ion channels, cardiac structural proteins, cardiogenesis, cardiac development, energy metabolism and genes related to neurohormal regulation, have been implicated in development of SCD (11).

Figure 1: The suggested pathophysiological mechanisms of sudden cardiac death
One of the proposed and, possibly unifying mechanism, is the attribution of SCD to the sympathetic stress response (1). This stress response is an adaptive mechanism that responds to aggressive stimuli to elicit behavioural (psychological) and physical (physiological) alterations through both the sympathetic-adrenal-medullary (SAM) and the hypothalamic-pituitary-adrenal (HPA) systems (32,33). The activation of these axes causes the secretion of norepinephrine and epinephrine (through SAM action), and glucocorticoids (through HPA action), to support the survival of different organs (34). Although this acute adaptability improves cardiac function for brief periods of time and is necessary for normal survival, prolonged activation of the sympathetic stress mechanism can have detrimental effects including ventricular remodelling and enhanced arrhythmogenesis (32,35).
A network of sympathetic neurons innervates cardiac tissue, including the SA node, AV node and the ventricles (36). Catecholamines are released from nerve terminals and act on α- and β-adrenergic receptors, causing an increase in heart rate (through action on SA node) and force of contraction (through action on ventricular muscle) (36). In response to acute sympathetic outflow, neuropeptide Y, a 36-amino acid polypeptide plentifully found in areas of the brain that regulate stress and emotional behaviour, is activated and inhibits the release of norepinephrine at the presynaptic level (37,38). On persistent sympathetic activation over a longer period of time, however, the activity of neuropeptide Y is significantly decreased, leading to a further increase in catecholamine release (37). Thus, it appears that a loss of modulatory action of neuropeptide Y on the sympathetic nerve terminal may play a critical role in elevating the levels of catecholamines in circulation due to prolonged stress.
An excessive amount of catecholamines has both direct and indirect consequences that may have deleterious impact on normal heart function (32,39). First, stimulation of α-adrenergic receptors by norepinephrine has been implicated to play a role in the progression of various cardiovascular diseases (40). Normal stimulation of α-adrenoceptors has a positive chronotropic effect; however, prolonged stimulation has been implicated in myocyte hypertrophy, myocardial ischemia, metabolic derangements and eventual cardiomyocyte apoptosis and the development of dilated cardiomyopathy (40-42). Despite this possible negative effect, neither the α-adrenoceptor density nor the positive inotropic effect of norepinephrine is distorted in failing human hearts, leading to the consideration that the α-adrenoceptor stimulation may be an adaptive mechanism in failing hearts (43). It should also be mentioned that stimulation of ß-adrenergic receptors by catecholamines activates adenylyl cyclase, causing the conversion of ATP to cAMP, a process that depletes high-energy phosphate stores (1,44). cAMP acts as a messenger molecule to activate protein kinase A (PKA), which phosphorylates voltage gated L-type calcium channels causing a Ca2+ influx into myocardial cells across the sarcolemma (1,45). PKA also induces phosphorylation of phospholamban and troponin, which impedes their normal lusitropic effect (32). The excessive inflow of calcium prompts the uncontrolled release of more Ca2+ from the sarcoplasmic reticulum (SR) via type 2 ryanodine receptors, which results in an intracellular Ca2+ overload (46,47). This adjustment in the calcium handling process of cardiomyocytes is known to increase the probability and occurrence of arrhythmias (Figure 2) including premature ventricular contractions, nonsustained ventricular tachycardia and ventricular fibrillation, thereby relating to SCD (1,48). The severity of these arrhythmias was shown to be dose dependent; thus, as the concentration of catecholamines increases, the chance of experiencing lethal ventricular fibrillation increases (49). Prolonged stimulation of α-adrenergic receptors may also result in coronary spasms resulting in myocardial ischemia and functional hypoxia, which may cause many of the underlying etiologies of SCD (1).

Figure 2: The mechanism by which excess catecholamines increase the occurrence of arrhythmias that may lead to sudden cardiac death. SL Sarcolemma; SR Sarcoplasmic reticulum
Furthermore and, possibly, most of the detrimental effects of the excessive release of catecholamines is due to the oxidation of surplus circulating catecholamines into aminochromes and the formation of reactive oxygen species (ROS) (Figure 3). Catecholamines become available for oxidation in the body when mechanisms (monoamineoxidase [MAO]) and catechol-o-methyl transferase [COMT]), which degrade them are saturated or impaired due to excessive catecholamines in the circulation or oxidative stress (50,51). This leads to the formation of aminochromes, which are known to deplete the high-energy phosphate stores, increase the intracellular calcium concentration leading to Ca2+ overload, cause lipid peroxidation, coronary artery spasms, arrhythmias, ventricular dysfunction and ultrastructural damage (46). This was shown on the perfusion of isolated rat hearts with oxidized isoproterenol, which generated notable morphological changes and contractile failure unlike its unoxidized precursor (39). Furthermore, aminochromes may induce certain defects leading to SCD in patients with pre-existing primary electrical abnormalities (39).

Figure 3: The cascade of events leading to the development of oxidative stress following a sustained stressful stimulus and the subsequent release of excess catecholamines. ROS Reactive oxygen species
The oxidation of circulating catecholamines is also known to produce oxyradicals (ie, ROS), which can easily react with a number of cellular structures and whose excess leads to the development of oxidative stress (39,51). Amplified oxidative stress results in dysfunction of normal electrical functioning and intracellular ion homeostasis in cardiac myocytes, making it widespread in arrhythmogenic cardiac conditions, including SCD (52). Along with the process of autooxidation of the excess circulating catecholamines, their interaction with MAO becomes an inherent enduring source of ROS in the heart during degradation of catecholamines, and the overstimulation of MAO results in the continuation of mitochondrial damage (53). Blockade of autophagic flux by MAO-A activity, and increased generation of ROS through MAO-B activity, lead to the build-up of damaged mitochondria and cell death (53). Detrimental effects of ROS generated in the heart include decline in contractile force, ultrastructural changes and arrhythmias, all of which may contribute to SCD (9). Different studies (49,54) aimed at testing the efficacy of antioxidants in preventing the catecholamine-induced arrhythmias support the postulation that it may be aminochromes and ROS rather than the epinephrine and norepinephrine themselves that play a major role in catecholamineinduced SCD. A marked decrease in the incidence of catecholamineinduced arrhythmias on pretreatment with antioxidants, such as N-acetyl-L-cysteine as well as vitamins A, C, and E, has been reported in the literature (49,54).
Activation of the HPA axis also contributes to catecholamineinduced SCD through the action of glucocorticoids, which are released from the adrenal cortex in response to a stressful stimulus and amplify the effects of sympathetic stimulation (32). The main action of glucocorticoids is the inhibition of extraneuronal uptake of catecholamines. However, other effects include increased synthesis and decreased degradation of catecholamines, directly increasing the concentration of circulating catecholamines (32). Furthermore, these hormones also have been shown to increase the total population, binding capability and affinity of β1-adrenergic receptors, further enhancing the effects of sympathetic stimulation (32).
Prevention and Treatment
Given the high rate of sudden death in event of cardiac arrest, the pursuit of preventive measures and effective treatment must be unremitting (15,55). Wearable and implantable cardiac defibrillators are one of the leading methods of prevention of SCD in HF patients and other high-risk populations, showing notable reduction of mortality in randomized clinical trials (10,58). Holding the capability to cardiovert, defibrillate, and pace the heart, these devices are often used as prophylactic treatment for patients at high risk for SCD caused by VF (1,12). More invasive treatment options include the use of percutaneous angioplasty, coronary interventions such as coronary bypass surgery for CAD patients, and the development of drug-eluting stents to be placed in diseased coronary arteries (1). Various pharmaceutical treatments are also available for SCD patients, the most obvious candidate being anti-arrhythmic drugs (10). Despite their theoretical benefit, evidence supporting the use of these drugs is sparse due to a lack of convincing studies to support their use for primary prevention of SCD (10). It should be noted that various antiplatelet agents, including sarpogrelate, a 5-HT2A antagonist, and acetylsalicylic acid have been shown to improve cardiac function and reduce the occurrence of ventricular arrhythmias related to SCD, indicating their potential use as prophylactic treatment of SCD (1,48).
Because high concentrations of circulating catecholamines are known to contribute to the development and progression of various cardiovascular diseases, therapeutic interventions concentrate on inhibition of sympathetic activity (56,59). This is typically achieved through the use of β-adrenoceptor blockers, which have been shown to decrease the risk for SCD by up to 31% in clinical trials (57). These drugs work by weakening the effect of the activation of β-adrenergic receptors by catecholamines, and have been shown to improve left ventricular function, slow cardiac remodelling and reduce mortality in HF patients (1,59,60). Although most of the beneficial effects of β-adrenoceptor blockers have typically been attributed to the lowering of heart rate in HF patients, β-blockers, such as metoprolol, propranolol and atenolol, have all been shown to depress high levels of plasma norepinephrine and epinephrine through effects on preterminal sympathetic nerve endings, further highlighting these agents for treatment of catecholamine-induced SCD (59,61). Similarly, blockade of α-adrenergic receptors by drugs, such as prazosin, has also been shown to have a similar effect, depressing elevated plasma norepinephrine levels (59). More invasive methods of attenuating the effects of sympathetic nervous system induced cardiac pathologies include renal and cardiac sympathetic denervation and increasing parasympathetic activity, which opposes the effects of sympathetic stimulation through vagal nerve stimulation. However, further studies are needed to assess the efficacy and long-term consequences of these treatments (56,62).
As outlined previously, deleterious effects of the oxidation products of catecholamines and their ability to induce arrhythmias leading to SCD, different antioxidants, such as N-acetyl-L-cysteine, vitamin A, vitamin C, and vitamin E, have been shown to attenuate the aminochrome and ROS induced cardiac structural damage and dysfunction (39,49,54). The beneficial effects in hypertension, ischemic heart disease, various cardiomyopathies, and heart failure have been reported to be due antioxidant therapy (49). N-acetyl-Lcysteine, vitamin C, and vitamin A have been shown to exert antiarrhythmic effects, whereas vitamin E has been demonstrated to reduce the incidence of premature ventricular contractions and exert antiapoptotic action (49,54,63). Due to their ability to lower plasma aminochrome levels and reduce the deleterious effects of oxidation products with respect to catecholamine induced arrhythmias and SCD, administration of antioxidants may be a promising treatment option for patients with different types of arrhythmia. However, because clinical evidence has not fully supported data from experimental studies, further evaluation of their effects is needed before drawing concrete conclusions (49,54).
Conclusions
SCD is one of the leading causes of death worldwide, and may be the manifestation of a large range of different cardiovascular abnormalities and diseases. Due to complications in the assessment for risk and diagnosis for SCD, development of a distinct set of guidelines for treatment for different causes and diseases is required, but it is only possible with further research in this field. Due to the possibility of many of the underlying comorbidities being caused by the sympathetic stress response and the release and oxidation of excess catecholamines, focus on this causation for SCD may provide insight to the main pathophysiological mechanism of this complex disease. Nonetheless, development of efficacious and widely applicable treatment options is necessary, with a combination of β-adrenoceptor blockers and antioxidants holding some merit in this field due to their ability to attenuate both the direct and indirect damaging effects of excess catecholamines.
Acknowledgments
Infrastructural support for this study was provided by the St Boniface Hospital Foundation, Winnipeg, Manitoba, Canada. AA has been supported by a grant from APPV (15-607) and VEGA (1/0271/16).
References
- Goyal V, Jassal DS, Dhalla NS. Pathophysiology and prevention of sudden cardiac death. Can J Physiol Pharmacol 2016;94:237-44.
- Stecker EC, Reinier K, Marijon E, et al. Public health burden of sudden cardiac death in the United States. Circ Arrhythm Electrophysiol 2014;7:212-7.
- Kuriachan VP, Sumner GL, Mitchell LB. Sudden cardiac death. Curr Probl Cardiol 2015;40:133-200.
- Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015;116:1887-906.
- Mitrani RD, Myerburg RJ. Ten advances defining sudden cardiac death. Trends Cardiovasc Med 2016;26:23-33.
- Saffitz JE, Corradi D. The electrical heart: 25 years of discovery in cardiac electrophysiology, arrhythmias and sudden death. Cardiovasc Pathol 2016;25:149-57.
- Gatzoulis KA, Tsiachris D, Arsenos P, Tousoulis D. Electrophysiologic testing guided risk stratification approach for sudden cardiac death beyond the left ventricular ejection fraction. World J Cardiol 2016;8:112-3.
- Maeda R, Minami Y, Haruki S, et al. Implantable cardioverter defibrillator therapy and sudden death risk stratification in hypertrophic cardiomyopathy patients with midventricular obstruction: A single-center experience. Int J Cardiol 2016;214:419-22.
- Al-Gobari M, El Khatib C, Pillon F, Gueyffier F. β-blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc Disord 2013;13:52.
- Weeks PA, Sieg A, Gass JA, Rajapreyar I. The role of pharmacotherapy in the prevention of sudden cardiac death in patients with heart failure. Heart Fail Rev 2016;21:415-31.
- Wei D, Tao L, Huang M. Genetic variations involved in sudden cardiac death and their associations and interactions. Heart Fail Rev 2016;21:401-14.
- Biton Y, Baman JR, Polonsky B. Roles and indications for use of implantable defibrillator and resynchronization therapy in the prevention of sudden cardiac death in heart failure. Heart Fail Rev 2016;21:433-46.
- Sun WP, Li CL, Guo JC, Zhang LX, Liu R, Zhang HB, Zhang L. Long-term efficacy of implantable cardiac resynchronization therapy plus defibrillator for primary prevention of sudden cardiac death in patients with mild heart failure: An updated meta-analysis. Heart Fail Rev 2016;21:447-53.
- Vaduganathan M, Patel RB, Shah SJ, Butler J. Sudden cardiac death in heart failure with preserved ejection fraction: A target for therapy? Heart Fail Rev 2016;21:455-62.
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and Stroke Statistics - 2016 Update: A Report From the American Heart Association. Circulation 2016; 133:e38-60.
- Israel CW. Mechanisms of sudden cardiac death. Indian Heart J 2014;66:S10-7.
- Tse G. Mechanisms of cardiac arrhythmias. J Arrhythm 2016;32:75-81.
- American Heart Association. Coronary artery disease. 2015 <www.heart.org/HEARTORG/Conditions/More/ MyHeartandStrokeNews/Coronary-Artery-Disease---Coronary- Heart-Disease_UCM_436416_Article.jsp#.V358z5MrL-Z> (Accessed July 24, 2016).
- Kim SS, Ko SM, Choi SI, Choi BH, Stillman AE. Sudden cardiac death from structural heart diseases in adults: imaging findings with cardiovascular computed tomography and magnetic resonance. Int J Cardiovasc Imaging. 2016; 32:21-43.
- Sara JD, Eleid MF, Gulati R, Holmes DR Jr. Sudden cardiac death from the perspective of coronary artery disease. Mayo Clin Proc. 2014; 89:1685-98.
- American Heart Association. What is cardiomyopathy in adults? <www.heart.org/HEARTORG/Conditions/More/Cardiomyopathy/ What-Is-Cardiomyopathy-in-Adults_UCM_444168_Article.jsp#. V359x5MrJfQ> (Accessed July 24, 2016).
- Kocovski L, Fernandes J. Sudden cardiac death: a modern pathology approach to hypertrophic cardiomyopathy. Arch Pathol Lab Med 2015;139:413-6.
- Te Riele AS, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: clinical challenges in a changing disease spectrum. Trends Cardiovasc Med 2015;25:191-8.
- Scardulla F, Rinaudo A, Pasta S, Scardulla C. Evaluation of ventricular wall stress and cardiac function in patients with dilated cardiomyopathy. Proc Inst Mech Eng H 2016;230:71-4.
- Goldberger JJ. Sudden cardiac death risk stratification in dilated cardiomyopathy: climbing the pyramid of knowledge. Circ Arrhythm Electrophysiol 2014; 7:1006-8.
- Basso C, Perazzolo Marra M, Rizzo S, De Lazzari M, Giorgi B, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation. 2015; 132:556-66.
- American Heart Association. Problem: Aortic valve stenosis. 2016; <www.heart.org/HEARTORG/Conditions/More/ HeartValveProblemsandDisease/Problem-Aortic-Valve-Stenosis_ UCM_450437_Article.jsp#.V3rAcpMrLPB>.
- American Heart Association. Problem: Mitral valve prolapse. 2016; <www.heart.org/HEARTORG/Conditions/More/ HeartValveProblemsandDisease/Problem-Mitral-Valve-Prolapse_ UCM_450441_Article.jsp#.V3BN35MrL-Y>
- Rayburn WF. Diagnosis and classification of diabetes mellitus: highlights from the American Diabetes Association. J Reprod Med 1997;42:585-6.
- Walker AM, Cubbon RM. Sudden cardiac death in patients with diabetes mellitus and chronic heart failure. Diab Vasc Dis Res 2015;12:228-33.
- Israel CW, Lee-Barkey YH. Sudden cardiac death in diabetes mellitus. Herz 2016;41:193-200.
- Adameova A, Abdellatif Y, Dhalla NS. Role of the excessive amounts of circulating catecholamines and glucocorticoids in stress- induced heart disease. Can J Physiol Pharmacol 2009;87:493-514.
- Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci 1998;851:311-35.
- Hannibal KE, Bishop MD. Chronic stress, cortisol dysfunction, and pain: A psychoneuroendocrine rationale for stress management in pain rehabilitation. Phys Ther. 2014;94:1816-25.
- Richard E. Klabunde. Sympathetic activation in heart failure. Cardiovascular Physiology Concepts. 2015 <www.cvphysiology.com/ Heart%20Failure/HF004.htm> (Accessed July 24, 2016).
- Wengrowski AM, Wang X, Tapa S, et al. Optogenetic release of norepinephrine from cardiac sympathetic neurons alters mechanical and electrical function. Cardiovasc Res 2015;105:143-50.
- Basu S, Sinha SK, Shao Q, Ganguly PK, Dhalla NS. Neuropeptide Y modulation of sympathetic activity in myocardial infarction. J Am Coll Cardiol 1996;27:1796-803.
- Sah R, Geracioti TD. Neuropeptide Y and posttraumatic stress disorder. Mol Psychiatry 2013;18:646-55.
- Dhalla NS, Adameova A, Kaur M. Role of catecholamine oxidation in sudden cardiac death. Fundam Clin Pharmacol 2010;24:539-46.
- Shannon R, Chaudhry M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am Heart J 2006;152:842-50.
- Landzberg JS, Parker JD, Gauthier DF, Colucci WS. Effects of myocardial alpha 1-adrenergic receptor stimulation and blockade on contractility in humans. Circulation 1991;84:1608-14.
- Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res 2000; 45:713-9.
- Babick A, Elimban V, Dhalla NS. Reversal of cardiac remodeling and subcellular defects by prazosin in heart failure due to myocardial infarction. J Clin Exp Cardiol 201;S5:009.
- Schömig A. Catecholamines in myocardial ischemia: Systemic and cardiac release. Circulation 1990; 82:II13-22.
- Hoang-Trong TM, Ullah A, Jafri MS. Calcium sparks in the heart: Dynamics and regulation. Res Rep Biol 2015; 6:203-214.
- Castellano M, Böhm M. The cardiac beta-adrenoceptor-mediated signaling pathway and its alterations in hypertensive heart disease. Hypertension 1997;29:715-22.
- Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 2015;112:11389-94.
- Barta J, Sanganalmath SK, Kumamoto H, Takeda N, Edes I, et al. Antiplatelet agents sarpogrelate and cilostazol affect experimentally- induced ventricular arrhythmias and mortality. Cardiovasc Toxicol 2008;8:127-35.
- Sethi R, Rehsia NS, Jindal K, Dhalla KS, Elimban V, et al. Antiarrhythmic effects of some antioxidant vitamins in rats injected with epinephrine. Cardiovasc Toxicol 2009; 9:177-84.
- Ganguly PK, Beamish RE, Dhalla NS. Catecholamine cardiotoxicity in pheochromocytoma. Am Heart J 1989;117:1399-400.
- Rouleau JL, Pitt B, Dhalla NS, Dhalla KS, et al. Prognostic importance of the oxidized product of catecholamines, adrenolutin, in patients with severe heart failure. Am Heart J 2003;145:926-32.
- Yang KC, Kyle JW, Makielski JC, Dudley SC Jr. Mechanisms of sudden cardiac death: Oxidants and metabolism. Circ Res 2015;116:1937-55.
- Santin Y, Sicard P, Vigneron F, et al. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal 2016;25:10-27.
- Sethi R, Adameova A, Dhalla KS, et al. Modification of epinephrine- induced arrhythmias by N-acetyl-L-cysteine and vitamin E. J Cardiovasc Pharmacol Ther 2009;14:134-42.
- Chugh SS, Reinier K. Predicting sudden death in the general population: another step, N terminal B-type natriuretic factor levels. Circulation 2009;119:2863-4.
- He B, Lu Z, He W, Huang B, Jiang H. Autonomic modulation by electrical stimulation of the parasympathetic nervous system: An emerging intervention for cardiovascular diseases. Cardiovasc Ther 2016;34:167-71.
- Al-Gobari M, El Khatib C, Pillon F, Gueyffier F. β-Blockers for the prevention of sudden cardiac death in heart failure patients: a meta- analysis of randomized controlled trials. BMC Cardiovasc Disord 2013;13:52.
- Piccini JP Sr, Allen LA, Kudenchuk PJ, et al. Wearable cardioverter- defibrillator therapy for the prevention of sudden cardiac death: A science advisory from the American Heart Association. Circulation 2016;133:1715-27.
- Babick A, Elimban V, Zieroth S, Dhalla NS. Reversal of cardiac dysfunction and subcellular alterations by metoprolol in heart failure due to myocardial infarction. J Cell Physiol 2013;228:2063-70.
- Doughty RN, Rodgers A, Sharpe N, MacMahon S. Effects of beta- blocker therapy on mortality in patients with heart failure: A systematic overview of randomized controlled trials. Eur Heart J 1997; 18:560-5.
- Machackova J, Sanganalmath SK, Barta J, Dhalla KS, Dhalla NS. Amelioration of cardiac remodeling in congestive heart failure by beta-adrenoceptor blockade is associated with depression in sympathetic activity. Cardiovasc Toxicol 2010;10:9-16.
- Huang B, Yu L, He B, Wang S, Lu Z, et al. Sympathetic denervation of heart and kidney induces similar effects on ventricular electrophysiological properties. EuroIntervention 2015;11:598-604.
- Kirshenbaum LA, Gupta M, Thomas TP, Singal PK. Antioxidant protection against adrenaline-induced arrhythmias in rats with chronic heart hypertrophy. Can J Cardiol 1990; 6:71-4.
- *Corresponding Author:
- Dr Naranjan S Dhalla
Institute of Cardiovascular Sciences, St Boniface Research Centre, 351 Tache Avenue, Winnipeg, Manitoba R2H 2A6
Tel: 204-235-3417, fax 204-237-0347
E-mail: nsdhalla@srbc.ca
This open-access article is distributed under the terms of the Creative Commons Attribution Non-Commercial License (CC BY-NC) (http:// creativecommons.org/licenses/by-nc/4.0/), which permits reuse, distribution and reproduction of the article, provided that the original work is properly cited and the reuse is restricted to noncommercial purposes. For commercial reuse, contact reprints@pulsus.com
Abstract
Sudden cardiac death (SCD) is a complex disease and represents one of the largest burdens to health care worldwide. It is known to be associated with various disease conditions, including but not limited to, cardiac arrhythmias, coronary artery disease, cardiomyopathies, valvular disorders and diabetes. Although much work is left to be done in uncovering the underlying mechanisms of SCD, response of the sympathetic nervous system to a prolonged stressful stimulus and the release of excessive amounts of catecholamines and their subsequent oxidation has potential to explain the regularly observed etiologies and provide a unified mechanism for the occurrence of SCD. Current possible treatments for patients at risk for SCD range from the implantation of cardioverter defibrillators to pharmaceutical interventions including anti-arrhythmic drugs, antiplatelet agents and β-adrenoceptor blockers. However, there are some studies suggesting the merit of prophylactic treatment using antioxidants such as vitamins A, C and E in preventing arrhythmias and consequent SCD. Overstimulation of the sympathetic stress response may result in SCD, and combination therapy with antioxidants and β-adrenoceptor blockers may be suitable for its prevention.
-Keywords
Antioxidants; ß-adrenoceptor blockers; Cardiac arrhythmias; Catecholamines; Sudden cardiac death; Sympathetic nervous system
Sudden cardiac death (SCD) is characterized as unforeseen failure of the heart due to cardiac arrhythmias, resulting in acute symptoms including palpitations, vomiting and fatigue, followed by severe symptoms such as rapid syncope, loss of breathing, loss of pulse, sudden collapse and, eventually, death (1,2). Most sudden deaths are related to cardiac issues, accounting for more than 15% to 20% of global deaths (300,000 to 400,000 in the United States, depending on the definition) per year (3,4). More than 50% of these deaths occur as the initial manifestation of an underlying heart disease, with cardiac arrest occurring within 1 h to 2 h of symptom onset, in what would normally be considered a low-risk individual (1,5). The underlying disease may result in either structural defects or electrical abnormalities, both of which can lead to serious ventricular arrhythmias and, eventually, SCD through multiple mechanisms (3). Occurrence of structural, electrical and metabolic changes, as well as electrolyte distribution alterations that can occur due to channelopathies, have been proposed to explain SCD. However, sustained sympathetic activation in response to a stressful stimulus and subsequent release of catecholamines and their oxidation has been suggested to be a unifying mechanism of SCD (1,3). Despite the fact that a large proportion of SCD victims exhibited coronary artery disease (CAD), irregular electrocardiographic patterns and ventricular remodelling due to myocardial infarction or vascular anomaly, risk stratification is challenging because the same pathophysiological indicators are present in many patients who have not experienced SCD (6,7). Aside from drugs modulating ion channels and drugs affecting the renin-angiotensin system, typical therapy of high-risk patients involves the use of drugs (ß-adrenoceptor blockers) that oppose the effects of catecholamines as well as the implantation of cardioverter defibrillators (1,8). Both the pathogenesis and the prevention/treatment of SCD have been extensively reviewed in the recent literature (9-14) and, based on these, the American College of Cardiology (ACC)/American Heart Association (AHA) as well as European Society of Cardiology (ESC) have updated their guidelines regarding this topic. The purpose of the present article is to briefly discuss the underlying etiologies, risk stratification methods and treatment strategies for SCD, and provide a detailed assessment of the mechanisms associated with SCD, mainly the release and subsequent oxidation of excessive catecholamines through the sympathetic stress response.
Etiology
According to the 2016 Heart Disease and Stroke Statistics published by the AHA, the incidence of (emergency medical services assessed) out-of-hospital sudden cardiac arrest is approximately 110.8 per 100,000 individuals (356,500 individuals per year) in the United States, and the incidence of in-hospital sudden cardiac arrest is approximately 209,000 (15). The average survival rate of patients who experience an out-of-hospital sudden cardiac arrest is 10.8% and, for those who experience an in-hospital sudden cardiac arrest is 18.4% (15). The majority of patients who experience SCD are adult, with <1% of SCDs occurring in individuals <35 years of age; the incidence of SCD is higher in men than in women (4). While channelopathies and cardiomyopathies, myocarditis and substance abuse are the major cause of SCD in young subjects, chronic degenerative diseases (CAD, valvular heart diseases and HF) predominate in the pathogenesis of the disease in older populations. Table 1 summarizes the incidence of SCD in the different cohorts indicated based on the most recent sudden cardiac arrest statistics (4,15).
Cardiac arrhythmias are the most common cause of SCD; these involve irregularity of electrical activity, the most common form associated with SCD being ventricular fibrillation (VF), which causes desynchronization of ventricular contractions (1,3). Ventricular arrhythmias are encountered in almost all SCD patients, 75% to 80% exhibiting VF, 10% to 15% exhibiting bradyarrhythmias and only 5% to 10% without indication of arrhythmia (16). This disruption in the order of the transmittance of the excitation wave (generated at the sinoatrial node) results in improper beating of the heart and, thus, incomplete pumping and circulation of blood (17). Arrhythmias in patients without structural disease/irregularities result from primary electrical abnormalities, including Wolff-Parkinson-White (WPW) syndrome, long-QT syndrome (LQTS), short-QT syndrome (SQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT) and bradycardias (3). These observations can form the basis for the use of defibrillators for the correction of arrhythmias and prevention of SCD. Becuase the incidence of SCD is dependent on the type of certain cardiac diseases, it is considered appropriate to discuss this aspect in some detail.
CAD and Cardiomyopathies
The most common cause of SCD is CAD, which develops as a result of damage or disease (typically due to plaque formation and inflammation) affecting the coronary arteries, which supply oxygenated blood and nutrients to the heart (18). CAD is the most common cause of SCD in adults >40 years of age, with atherosclerotic CAD accounting for 75% to 80% of the cases in this cohort (19). It also needs to be emphasized that most of these SCD cases involve seemingly normal adults with no symptoms or history of CAD; therefore, some caution should be exercised for explaining the mechanisms of SCD in patients with CAD (20).
Similar to ischemic cardiomyopathy, both hypertrophic and dilated cardiomyopathies are conditions that involve abnormalities of the heart muscle, diminishing the ability of the heart to pump blood or maintain normal electrical rhythm (21). Cardiomyopathies commonly associated with SCD include hypertrophic cardiomyopathy, arrhythmogenic right ventricular dysplasia, and dilated cardiomyopathy (1). Hypertrophic cardiomyopathy is a heritable autosomal dominant disorder and is the most common cause of SCD in young athletes. It involves abnormal growth of cardiomyocytes, causing thickening of the heart’s muscular walls and cardiomyocyte misalignment (myocardial disarray), causing them to become nonfunctional (1,22).Arrhythmogenic right ventricular dysplasia is another inherited cardiomyopathy that involves predominantly right ventricular fibro-fatty replacement of cardiomyocytes, predisposing patients to lethal arrhythmias associated with SCD (1,23). Finally, dilated cardiomyopathy involves both left ventricular dilation and systolic dysfunction, resulting in cardiac remodelling and, consequently HF, and is the second leading cause of SCD after CAD (24,25).
Valvular disorders
Valvular disorders, including aortic stenosis and mitral valve prolapse, can also manifest ventricular arrhythmias, prompting SCD (1,26). Aortic stenosis is the narrowing of aortic valve outlet of the left ventricle, restricting blood flow to the aorta, causing shortness of breath or breathlessness, angina, palpitations, heart murmurs and syncope (27). If left untreated, a decrease in coronary blood flow and overall decreased cardiac output due to acute left ventricular failure can result in SCD (1). On the other hand, mitral valve prolapse is caused by uneven closure of the mitral valve, causing it to protrude (prolapse) into the left atrium, resulting in the backward leak of small amounts of blood (28). Even in the absence of notable hemodynamic impairment, mitral valve prolapse can produce ventricular arrhythmias and SCD (26).
Diabetes mellitus
Diabetes mellitus comprises a collection of metabolic diseases, which cause hyperglycemia rooted in faults in either insulin secretion due to pancreatic β-cell dysfunction as well as abnormalities resulting in resistance to insulin action, or both, affecting >382 million individuals worldwide (29,30). Acute symptoms of hyperglycemia include polyuria, polydipsia, weight loss and blurred vision (29). Long-term induction of hyperglycemia is linked with the impairment of growth, increased susceptibility to infections, and damage and dysfunction of various organs, mainly the eyes, kidneys, heart and blood vessels, as well as nerves (29). The risk for SCD is significantly increased in diabetes mellitus patients, with large-group studies indicating a two to four times increase in the incidence of SCD compared with the normal population (30). SCD is one of the most prominent causes of death in patients with diabetes, accounting for approximately 50% of deaths due to cardiovascular causes in this cohort (30,31).
Mechanisms of SCD
Several mechanisms have been proposed in an attempt to explain SCD.These include the presence of vascular anomaly and mainly cardiac structural, electrical, metabolic and electrolyte abnormalities, as well as genetic variations (Figure 1). Structural heart disease can lead to the development of SCD, giving rise to the postulation that SCD is caused solely by structural abnormalities in CAD, ischemic cardiomyopathies, congenital heart disease, valvular heart diseases and various cardiomyopathies (3). Electrophysiological/electrical abnormalities have also been considered as a possible mechanism for SCD, accrediting SCD to the arrhythmogenic primary electrical abnormalities including WPW, LQTS, SQTS, BrS and CPVT. The appearance of focal activity, such as enhanced automaticity and triggered activity or reentry, which can occur due to an anatomical or functional obstacle, has also been suggested to result in SCD (3,17). Lifethreatening arrhythmias, which can lead to SCD, are also known to be associated with electrolyte imbalances, such as hypokalemia, but also hyperkalemia and hypomagnesemia, metabolic abnormalities, severe inflammation and endocrine disorders (hyper- and hypothyroidism) (3). More recently, several genetic variations, including those affecting ion channels, cardiac structural proteins, cardiogenesis, cardiac development, energy metabolism and genes related to neurohormal regulation, have been implicated in development of SCD (11).
Figure 1: The suggested pathophysiological mechanisms of sudden cardiac death
One of the proposed and, possibly unifying mechanism, is the attribution of SCD to the sympathetic stress response (1). This stress response is an adaptive mechanism that responds to aggressive stimuli to elicit behavioural (psychological) and physical (physiological) alterations through both the sympathetic-adrenal-medullary (SAM) and the hypothalamic-pituitary-adrenal (HPA) systems (32,33). The activation of these axes causes the secretion of norepinephrine and epinephrine (through SAM action), and glucocorticoids (through HPA action), to support the survival of different organs (34). Although this acute adaptability improves cardiac function for brief periods of time and is necessary for normal survival, prolonged activation of the sympathetic stress mechanism can have detrimental effects including ventricular remodelling and enhanced arrhythmogenesis (32,35).
A network of sympathetic neurons innervates cardiac tissue, including the SA node, AV node and the ventricles (36). Catecholamines are released from nerve terminals and act on α- and β-adrenergic receptors, causing an increase in heart rate (through action on SA node) and force of contraction (through action on ventricular muscle) (36). In response to acute sympathetic outflow, neuropeptide Y, a 36-amino acid polypeptide plentifully found in areas of the brain that regulate stress and emotional behaviour, is activated and inhibits the release of norepinephrine at the presynaptic level (37,38). On persistent sympathetic activation over a longer period of time, however, the activity of neuropeptide Y is significantly decreased, leading to a further increase in catecholamine release (37). Thus, it appears that a loss of modulatory action of neuropeptide Y on the sympathetic nerve terminal may play a critical role in elevating the levels of catecholamines in circulation due to prolonged stress.
An excessive amount of catecholamines has both direct and indirect consequences that may have deleterious impact on normal heart function (32,39). First, stimulation of α-adrenergic receptors by norepinephrine has been implicated to play a role in the progression of various cardiovascular diseases (40). Normal stimulation of α-adrenoceptors has a positive chronotropic effect; however, prolonged stimulation has been implicated in myocyte hypertrophy, myocardial ischemia, metabolic derangements and eventual cardiomyocyte apoptosis and the development of dilated cardiomyopathy (40-42). Despite this possible negative effect, neither the α-adrenoceptor density nor the positive inotropic effect of norepinephrine is distorted in failing human hearts, leading to the consideration that the α-adrenoceptor stimulation may be an adaptive mechanism in failing hearts (43). It should also be mentioned that stimulation of ß-adrenergic receptors by catecholamines activates adenylyl cyclase, causing the conversion of ATP to cAMP, a process that depletes high-energy phosphate stores (1,44). cAMP acts as a messenger molecule to activate protein kinase A (PKA), which phosphorylates voltage gated L-type calcium channels causing a Ca2+ influx into myocardial cells across the sarcolemma (1,45). PKA also induces phosphorylation of phospholamban and troponin, which impedes their normal lusitropic effect (32). The excessive inflow of calcium prompts the uncontrolled release of more Ca2+ from the sarcoplasmic reticulum (SR) via type 2 ryanodine receptors, which results in an intracellular Ca2+ overload (46,47). This adjustment in the calcium handling process of cardiomyocytes is known to increase the probability and occurrence of arrhythmias (Figure 2) including premature ventricular contractions, nonsustained ventricular tachycardia and ventricular fibrillation, thereby relating to SCD (1,48). The severity of these arrhythmias was shown to be dose dependent; thus, as the concentration of catecholamines increases, the chance of experiencing lethal ventricular fibrillation increases (49). Prolonged stimulation of α-adrenergic receptors may also result in coronary spasms resulting in myocardial ischemia and functional hypoxia, which may cause many of the underlying etiologies of SCD (1).
Figure 2: The mechanism by which excess catecholamines increase the occurrence of arrhythmias that may lead to sudden cardiac death. SL Sarcolemma; SR Sarcoplasmic reticulum
Furthermore and, possibly, most of the detrimental effects of the excessive release of catecholamines is due to the oxidation of surplus circulating catecholamines into aminochromes and the formation of reactive oxygen species (ROS) (Figure 3). Catecholamines become available for oxidation in the body when mechanisms (monoamineoxidase [MAO]) and catechol-o-methyl transferase [COMT]), which degrade them are saturated or impaired due to excessive catecholamines in the circulation or oxidative stress (50,51). This leads to the formation of aminochromes, which are known to deplete the high-energy phosphate stores, increase the intracellular calcium concentration leading to Ca2+ overload, cause lipid peroxidation, coronary artery spasms, arrhythmias, ventricular dysfunction and ultrastructural damage (46). This was shown on the perfusion of isolated rat hearts with oxidized isoproterenol, which generated notable morphological changes and contractile failure unlike its unoxidized precursor (39). Furthermore, aminochromes may induce certain defects leading to SCD in patients with pre-existing primary electrical abnormalities (39).
Figure 3: The cascade of events leading to the development of oxidative stress following a sustained stressful stimulus and the subsequent release of excess catecholamines. ROS Reactive oxygen species
The oxidation of circulating catecholamines is also known to produce oxyradicals (ie, ROS), which can easily react with a number of cellular structures and whose excess leads to the development of oxidative stress (39,51). Amplified oxidative stress results in dysfunction of normal electrical functioning and intracellular ion homeostasis in cardiac myocytes, making it widespread in arrhythmogenic cardiac conditions, including SCD (52). Along with the process of autooxidation of the excess circulating catecholamines, their interaction with MAO becomes an inherent enduring source of ROS in the heart during degradation of catecholamines, and the overstimulation of MAO results in the continuation of mitochondrial damage (53). Blockade of autophagic flux by MAO-A activity, and increased generation of ROS through MAO-B activity, lead to the build-up of damaged mitochondria and cell death (53). Detrimental effects of ROS generated in the heart include decline in contractile force, ultrastructural changes and arrhythmias, all of which may contribute to SCD (9). Different studies (49,54) aimed at testing the efficacy of antioxidants in preventing the catecholamine-induced arrhythmias support the postulation that it may be aminochromes and ROS rather than the epinephrine and norepinephrine themselves that play a major role in catecholamineinduced SCD. A marked decrease in the incidence of catecholamineinduced arrhythmias on pretreatment with antioxidants, such as N-acetyl-L-cysteine as well as vitamins A, C, and E, has been reported in the literature (49,54).
Activation of the HPA axis also contributes to catecholamineinduced SCD through the action of glucocorticoids, which are released from the adrenal cortex in response to a stressful stimulus and amplify the effects of sympathetic stimulation (32). The main action of glucocorticoids is the inhibition of extraneuronal uptake of catecholamines. However, other effects include increased synthesis and decreased degradation of catecholamines, directly increasing the concentration of circulating catecholamines (32). Furthermore, these hormones also have been shown to increase the total population, binding capability and affinity of β1-adrenergic receptors, further enhancing the effects of sympathetic stimulation (32).
Prevention and Treatment
Given the high rate of sudden death in event of cardiac arrest, the pursuit of preventive measures and effective treatment must be unremitting (15,55). Wearable and implantable cardiac defibrillators are one of the leading methods of prevention of SCD in HF patients and other high-risk populations, showing notable reduction of mortality in randomized clinical trials (10,58). Holding the capability to cardiovert, defibrillate, and pace the heart, these devices are often used as prophylactic treatment for patients at high risk for SCD caused by VF (1,12). More invasive treatment options include the use of percutaneous angioplasty, coronary interventions such as coronary bypass surgery for CAD patients, and the development of drug-eluting stents to be placed in diseased coronary arteries (1). Various pharmaceutical treatments are also available for SCD patients, the most obvious candidate being anti-arrhythmic drugs (10). Despite their theoretical benefit, evidence supporting the use of these drugs is sparse due to a lack of convincing studies to support their use for primary prevention of SCD (10). It should be noted that various antiplatelet agents, including sarpogrelate, a 5-HT2A antagonist, and acetylsalicylic acid have been shown to improve cardiac function and reduce the occurrence of ventricular arrhythmias related to SCD, indicating their potential use as prophylactic treatment of SCD (1,48).
Because high concentrations of circulating catecholamines are known to contribute to the development and progression of various cardiovascular diseases, therapeutic interventions concentrate on inhibition of sympathetic activity (56,59). This is typically achieved through the use of β-adrenoceptor blockers, which have been shown to decrease the risk for SCD by up to 31% in clinical trials (57). These drugs work by weakening the effect of the activation of β-adrenergic receptors by catecholamines, and have been shown to improve left ventricular function, slow cardiac remodelling and reduce mortality in HF patients (1,59,60). Although most of the beneficial effects of β-adrenoceptor blockers have typically been attributed to the lowering of heart rate in HF patients, β-blockers, such as metoprolol, propranolol and atenolol, have all been shown to depress high levels of plasma norepinephrine and epinephrine through effects on preterminal sympathetic nerve endings, further highlighting these agents for treatment of catecholamine-induced SCD (59,61). Similarly, blockade of α-adrenergic receptors by drugs, such as prazosin, has also been shown to have a similar effect, depressing elevated plasma norepinephrine levels (59). More invasive methods of attenuating the effects of sympathetic nervous system induced cardiac pathologies include renal and cardiac sympathetic denervation and increasing parasympathetic activity, which opposes the effects of sympathetic stimulation through vagal nerve stimulation. However, further studies are needed to assess the efficacy and long-term consequences of these treatments (56,62).
As outlined previously, deleterious effects of the oxidation products of catecholamines and their ability to induce arrhythmias leading to SCD, different antioxidants, such as N-acetyl-L-cysteine, vitamin A, vitamin C, and vitamin E, have been shown to attenuate the aminochrome and ROS induced cardiac structural damage and dysfunction (39,49,54). The beneficial effects in hypertension, ischemic heart disease, various cardiomyopathies, and heart failure have been reported to be due antioxidant therapy (49). N-acetyl-Lcysteine, vitamin C, and vitamin A have been shown to exert antiarrhythmic effects, whereas vitamin E has been demonstrated to reduce the incidence of premature ventricular contractions and exert antiapoptotic action (49,54,63). Due to their ability to lower plasma aminochrome levels and reduce the deleterious effects of oxidation products with respect to catecholamine induced arrhythmias and SCD, administration of antioxidants may be a promising treatment option for patients with different types of arrhythmia. However, because clinical evidence has not fully supported data from experimental studies, further evaluation of their effects is needed before drawing concrete conclusions (49,54).
Conclusions
SCD is one of the leading causes of death worldwide, and may be the manifestation of a large range of different cardiovascular abnormalities and diseases. Due to complications in the assessment for risk and diagnosis for SCD, development of a distinct set of guidelines for treatment for different causes and diseases is required, but it is only possible with further research in this field. Due to the possibility of many of the underlying comorbidities being caused by the sympathetic stress response and the release and oxidation of excess catecholamines, focus on this causation for SCD may provide insight to the main pathophysiological mechanism of this complex disease. Nonetheless, development of efficacious and widely applicable treatment options is necessary, with a combination of β-adrenoceptor blockers and antioxidants holding some merit in this field due to their ability to attenuate both the direct and indirect damaging effects of excess catecholamines.
Acknowledgments
Infrastructural support for this study was provided by the St Boniface Hospital Foundation, Winnipeg, Manitoba, Canada. AA has been supported by a grant from APPV (15-607) and VEGA (1/0271/16).
References
- Goyal V, Jassal DS, Dhalla NS. Pathophysiology and prevention of sudden cardiac death. Can J Physiol Pharmacol 2016;94:237-44.
- Stecker EC, Reinier K, Marijon E, et al. Public health burden of sudden cardiac death in the United States. Circ Arrhythm Electrophysiol 2014;7:212-7.
- Kuriachan VP, Sumner GL, Mitchell LB. Sudden cardiac death. Curr Probl Cardiol 2015;40:133-200.
- Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015;116:1887-906.
- Mitrani RD, Myerburg RJ. Ten advances defining sudden cardiac death. Trends Cardiovasc Med 2016;26:23-33.
- Saffitz JE, Corradi D. The electrical heart: 25 years of discovery in cardiac electrophysiology, arrhythmias and sudden death. Cardiovasc Pathol 2016;25:149-57.
- Gatzoulis KA, Tsiachris D, Arsenos P, Tousoulis D. Electrophysiologic testing guided risk stratification approach for sudden cardiac death beyond the left ventricular ejection fraction. World J Cardiol 2016;8:112-3.
- Maeda R, Minami Y, Haruki S, et al. Implantable cardioverter defibrillator therapy and sudden death risk stratification in hypertrophic cardiomyopathy patients with midventricular obstruction: A single-center experience. Int J Cardiol 2016;214:419-22.
- Al-Gobari M, El Khatib C, Pillon F, Gueyffier F. β-blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc Disord 2013;13:52.
- Weeks PA, Sieg A, Gass JA, Rajapreyar I. The role of pharmacotherapy in the prevention of sudden cardiac death in patients with heart failure. Heart Fail Rev 2016;21:415-31.
- Wei D, Tao L, Huang M. Genetic variations involved in sudden cardiac death and their associations and interactions. Heart Fail Rev 2016;21:401-14.
- Biton Y, Baman JR, Polonsky B. Roles and indications for use of implantable defibrillator and resynchronization therapy in the prevention of sudden cardiac death in heart failure. Heart Fail Rev 2016;21:433-46.
- Sun WP, Li CL, Guo JC, Zhang LX, Liu R, Zhang HB, Zhang L. Long-term efficacy of implantable cardiac resynchronization therapy plus defibrillator for primary prevention of sudden cardiac death in patients with mild heart failure: An updated meta-analysis. Heart Fail Rev 2016;21:447-53.
- Vaduganathan M, Patel RB, Shah SJ, Butler J. Sudden cardiac death in heart failure with preserved ejection fraction: A target for therapy? Heart Fail Rev 2016;21:455-62.
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart Disease and Stroke Statistics - 2016 Update: A Report From the American Heart Association. Circulation 2016; 133:e38-60.
- Israel CW. Mechanisms of sudden cardiac death. Indian Heart J 2014;66:S10-7.
- Tse G. Mechanisms of cardiac arrhythmias. J Arrhythm 2016;32:75-81.
- American Heart Association. Coronary artery disease. 2015 <www.heart.org/HEARTORG/Conditions/More/ MyHeartandStrokeNews/Coronary-Artery-Disease---Coronary- Heart-Disease_UCM_436416_Article.jsp#.V358z5MrL-Z> (Accessed July 24, 2016).
- Kim SS, Ko SM, Choi SI, Choi BH, Stillman AE. Sudden cardiac death from structural heart diseases in adults: imaging findings with cardiovascular computed tomography and magnetic resonance. Int J Cardiovasc Imaging. 2016; 32:21-43.
- Sara JD, Eleid MF, Gulati R, Holmes DR Jr. Sudden cardiac death from the perspective of coronary artery disease. Mayo Clin Proc. 2014; 89:1685-98.
- American Heart Association. What is cardiomyopathy in adults? <www.heart.org/HEARTORG/Conditions/More/Cardiomyopathy/ What-Is-Cardiomyopathy-in-Adults_UCM_444168_Article.jsp#. V359x5MrJfQ> (Accessed July 24, 2016).
- Kocovski L, Fernandes J. Sudden cardiac death: a modern pathology approach to hypertrophic cardiomyopathy. Arch Pathol Lab Med 2015;139:413-6.
- Te Riele AS, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: clinical challenges in a changing disease spectrum. Trends Cardiovasc Med 2015;25:191-8.
- Scardulla F, Rinaudo A, Pasta S, Scardulla C. Evaluation of ventricular wall stress and cardiac function in patients with dilated cardiomyopathy. Proc Inst Mech Eng H 2016;230:71-4.
- Goldberger JJ. Sudden cardiac death risk stratification in dilated cardiomyopathy: climbing the pyramid of knowledge. Circ Arrhythm Electrophysiol 2014; 7:1006-8.
- Basso C, Perazzolo Marra M, Rizzo S, De Lazzari M, Giorgi B, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation. 2015; 132:556-66.
- American Heart Association. Problem: Aortic valve stenosis. 2016; <www.heart.org/HEARTORG/Conditions/More/ HeartValveProblemsandDisease/Problem-Aortic-Valve-Stenosis_ UCM_450437_Article.jsp#.V3rAcpMrLPB>.
- American Heart Association. Problem: Mitral valve prolapse. 2016; <www.heart.org/HEARTORG/Conditions/More/ HeartValveProblemsandDisease/Problem-Mitral-Valve-Prolapse_ UCM_450441_Article.jsp#.V3BN35MrL-Y>
- Rayburn WF. Diagnosis and classification of diabetes mellitus: highlights from the American Diabetes Association. J Reprod Med 1997;42:585-6.
- Walker AM, Cubbon RM. Sudden cardiac death in patients with diabetes mellitus and chronic heart failure. Diab Vasc Dis Res 2015;12:228-33.
- Israel CW, Lee-Barkey YH. Sudden cardiac death in diabetes mellitus. Herz 2016;41:193-200.
- Adameova A, Abdellatif Y, Dhalla NS. Role of the excessive amounts of circulating catecholamines and glucocorticoids in stress- induced heart disease. Can J Physiol Pharmacol 2009;87:493-514.
- Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci 1998;851:311-35.
- Hannibal KE, Bishop MD. Chronic stress, cortisol dysfunction, and pain: A psychoneuroendocrine rationale for stress management in pain rehabilitation. Phys Ther. 2014;94:1816-25.
- Richard E. Klabunde. Sympathetic activation in heart failure. Cardiovascular Physiology Concepts. 2015 <www.cvphysiology.com/ Heart%20Failure/HF004.htm> (Accessed July 24, 2016).
- Wengrowski AM, Wang X, Tapa S, et al. Optogenetic release of norepinephrine from cardiac sympathetic neurons alters mechanical and electrical function. Cardiovasc Res 2015;105:143-50.
- Basu S, Sinha SK, Shao Q, Ganguly PK, Dhalla NS. Neuropeptide Y modulation of sympathetic activity in myocardial infarction. J Am Coll Cardiol 1996;27:1796-803.
- Sah R, Geracioti TD. Neuropeptide Y and posttraumatic stress disorder. Mol Psychiatry 2013;18:646-55.
- Dhalla NS, Adameova A, Kaur M. Role of catecholamine oxidation in sudden cardiac death. Fundam Clin Pharmacol 2010;24:539-46.
- Shannon R, Chaudhry M. Effect of alpha1-adrenergic receptors in cardiac pathophysiology. Am Heart J 2006;152:842-50.
- Landzberg JS, Parker JD, Gauthier DF, Colucci WS. Effects of myocardial alpha 1-adrenergic receptor stimulation and blockade on contractility in humans. Circulation 1991;84:1608-14.
- Singh K, Communal C, Sawyer DB, Colucci WS. Adrenergic regulation of myocardial apoptosis. Cardiovasc Res 2000; 45:713-9.
- Babick A, Elimban V, Dhalla NS. Reversal of cardiac remodeling and subcellular defects by prazosin in heart failure due to myocardial infarction. J Clin Exp Cardiol 201;S5:009.
- Schömig A. Catecholamines in myocardial ischemia: Systemic and cardiac release. Circulation 1990; 82:II13-22.
- Hoang-Trong TM, Ullah A, Jafri MS. Calcium sparks in the heart: Dynamics and regulation. Res Rep Biol 2015; 6:203-214.
- Castellano M, Böhm M. The cardiac beta-adrenoceptor-mediated signaling pathway and its alterations in hypertensive heart disease. Hypertension 1997;29:715-22.
- Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 2015;112:11389-94.
- Barta J, Sanganalmath SK, Kumamoto H, Takeda N, Edes I, et al. Antiplatelet agents sarpogrelate and cilostazol affect experimentally- induced ventricular arrhythmias and mortality. Cardiovasc Toxicol 2008;8:127-35.
- Sethi R, Rehsia NS, Jindal K, Dhalla KS, Elimban V, et al. Antiarrhythmic effects of some antioxidant vitamins in rats injected with epinephrine. Cardiovasc Toxicol 2009; 9:177-84.
- Ganguly PK, Beamish RE, Dhalla NS. Catecholamine cardiotoxicity in pheochromocytoma. Am Heart J 1989;117:1399-400.
- Rouleau JL, Pitt B, Dhalla NS, Dhalla KS, et al. Prognostic importance of the oxidized product of catecholamines, adrenolutin, in patients with severe heart failure. Am Heart J 2003;145:926-32.
- Yang KC, Kyle JW, Makielski JC, Dudley SC Jr. Mechanisms of sudden cardiac death: Oxidants and metabolism. Circ Res 2015;116:1937-55.
- Santin Y, Sicard P, Vigneron F, et al. Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Antioxid Redox Signal 2016;25:10-27.
- Sethi R, Adameova A, Dhalla KS, et al. Modification of epinephrine- induced arrhythmias by N-acetyl-L-cysteine and vitamin E. J Cardiovasc Pharmacol Ther 2009;14:134-42.
- Chugh SS, Reinier K. Predicting sudden death in the general population: another step, N terminal B-type natriuretic factor levels. Circulation 2009;119:2863-4.
- He B, Lu Z, He W, Huang B, Jiang H. Autonomic modulation by electrical stimulation of the parasympathetic nervous system: An emerging intervention for cardiovascular diseases. Cardiovasc Ther 2016;34:167-71.
- Al-Gobari M, El Khatib C, Pillon F, Gueyffier F. β-Blockers for the prevention of sudden cardiac death in heart failure patients: a meta- analysis of randomized controlled trials. BMC Cardiovasc Disord 2013;13:52.
- Piccini JP Sr, Allen LA, Kudenchuk PJ, et al. Wearable cardioverter- defibrillator therapy for the prevention of sudden cardiac death: A science advisory from the American Heart Association. Circulation 2016;133:1715-27.
- Babick A, Elimban V, Zieroth S, Dhalla NS. Reversal of cardiac dysfunction and subcellular alterations by metoprolol in heart failure due to myocardial infarction. J Cell Physiol 2013;228:2063-70.
- Doughty RN, Rodgers A, Sharpe N, MacMahon S. Effects of beta- blocker therapy on mortality in patients with heart failure: A systematic overview of randomized controlled trials. Eur Heart J 1997; 18:560-5.
- Machackova J, Sanganalmath SK, Barta J, Dhalla KS, Dhalla NS. Amelioration of cardiac remodeling in congestive heart failure by beta-adrenoceptor blockade is associated with depression in sympathetic activity. Cardiovasc Toxicol 2010;10:9-16.
- Huang B, Yu L, He B, Wang S, Lu Z, et al. Sympathetic denervation of heart and kidney induces similar effects on ventricular electrophysiological properties. EuroIntervention 2015;11:598-604.
- Kirshenbaum LA, Gupta M, Thomas TP, Singal PK. Antioxidant protection against adrenaline-induced arrhythmias in rats with chronic heart hypertrophy. Can J Cardiol 1990; 6:71-4.